
RustQC: 60x Faster RNA-seq Quality Control
RustQC is a Rust reimplementation of 15 RNA-seq QC tools consolidated into a single binary. One pass through the BAM file gives functionally identical outputs, but with >60x faster run time and drastically reduced I/O. Built with AI by a developer who has never written a line of Rust (me, Phil).
View the DocumentationIn a companion post, we introduced rewrites.bio: a set of principles for responsibly rewriting bioinformatics tools with AI. RustQC is where those principles came from. It's the project that convinced us the community needs that conversation.

Wall-clock execution time for RNA-Seq QC on a full-size dataset (~186M paired-end reads, GRCh38). Timings from an nf-core/rnaseq pipeline run on AWS.
The QC Bottleneck in nf-core/rnaseq
The QC subworkflow in nf-core/rnaseq runs 15 separate tools: dupRadar, featureCounts, eight RSeQC modules, preseq, three samtools commands, and Qualimap. Each one solves a different analytical need, many have been part of the pipeline since I first started working on it 10 years ago (RSeQC was the second commit). Together they've shaped how the field thinks about RNA-Seq quality; they've been cited thousands of times and are taught in genomics courses worldwide.
Each of these tools was built to solve a specific problem, and they do that well. But when you assemble them into a pipeline, they start to add up - each tool reads the same BAM file, pulls its own container image, and stages gigabytes of data into its work directory. On a full-size paired-end human RNA-seq dataset (~186 million reads, GRCh38), the combined sequential wall time is over 15 hours per sample. Some of the tools are fragile at scale: RSeQC's TIN calculation and preseq are both disabled by default in nf-core/rnaseq because they fail frequently, and TIN alone takes nearly 10 hours to run.
On cloud infrastructure, these problems compound. Long-running tasks increase the chance of spot instance reclamation. A 10-hour TIN job can often run for 8 hours, get reclaimed, and have to restart from scratch. At the scale nf-core/rnaseq operates (over 20,000 repository clones from 1,679 unique users in a recent two-week period), these add up to millions of CPU-hours and thousands pipeline runs that need manual intervention. Not because the tools are bad, but because no individual tool was designed with the full pipeline context in mind.
From dupRadar to RustQC
RustQC started as a bit of a tongue-in-cheek experiment. dupRadar is one of the tools that causes the most pipeline failures in nf-core/rnaseq, so I prompted Claude Code to rewrite it in Rust, using Seqera AI as a subagent to fetch logs, metrics and results from a test run on Seqera Platform using the Seqera MCP. I left the terminal running in the background and a short while later, it had put together a Rust package. My post to our Slack thread read “I did a rust rewrite over lunch. Testing it now”. This was followed a little later by another message: “I realize that I might have accidentally also made a super fast Rust implementation of featureCounts in the process of doing this”. I found that my new tool not only compiled, but produced fairly solid looking outputs. I hadn't written a line of code, I've never written any Rust in my life, but LLMs had now reached a point that they could fully execute a complex rewrite like this with limited Human input.
If Claude could do this with dupRadar I thought, what else could it do? I turned to Seqera AI again and asked it to analyze the nf-core/rnaseq logs to identify other candidates for a rewrite. By the end of that first weekend, I had a functional beta of a new tool that covered 15 different QC tools from the pipeline. Doing all of this in a single tool made execution very efficient - all metrics are calculated in a single pass over the input BAM file, and I could steer the LLM to inspect its own code and suggest efficiency improvements. Nextflow made the validation steps straightforward: run the nf-core/rnaseq pipeline with the original tools, run it with RustQC, compare the outputs. I used Seqera AI to modify a fork of the rnaseq pipeline to do this, and also to run the benchmarks and analyze the results.
What RustQC Does
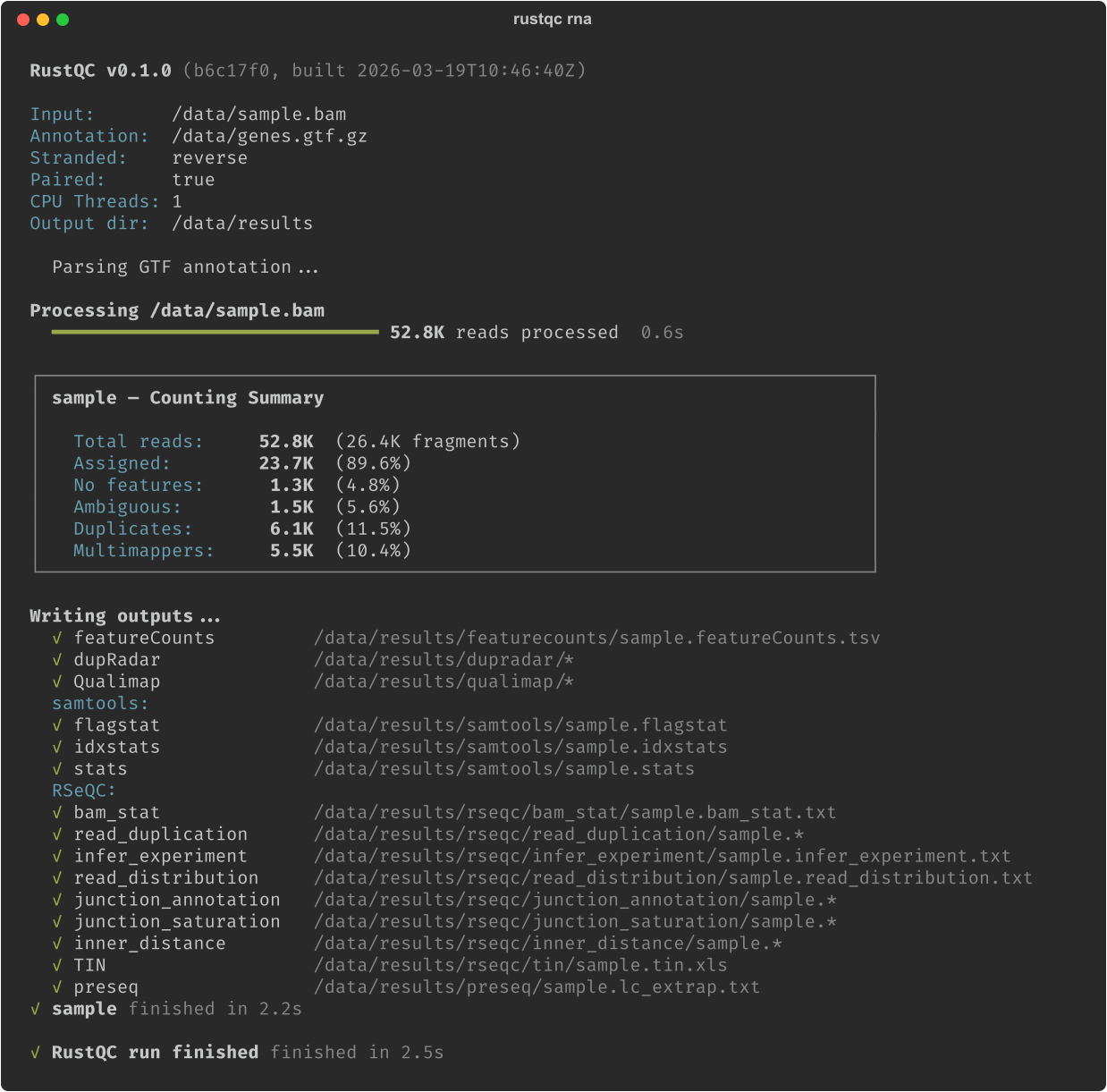
RustQC is a single compiled Rust binary that reimplements dupRadar, featureCounts, eight RSeQC modules (including TIN), preseq lc_extrap, samtools flagstat/idxstats/stats, and Qualimap RNA-Seq. It runs all of them in one pass through the BAM file.
On the same full-size dataset (~186M paired-end reads, GRCh38), the 15 original tools require over 15 hours of sequential wall time. RustQC produces all outputs in about 15 minutes: a greater than 60x speedup. The speedup comes from three main things:
- →The traditional workflow decompresses the same BAM file fifteen times independently. RustQC decodes it once and dispatches each aligned read to all analyses at the same time.
- →RustQC removes the need for certain preprocessing steps required by the original tools: Qualimap, for instance, needs a name-sorted BAM as input, adding over 12 minutes of upstream sorting in a separate process with its own tasks and intermediate files. RustQC works on the coordinate-sorted aligner output directly.
- →Many of the QC tools are single-threaded, but RustQC is able to parallelise work across chromosomes, making use of multi-core machines.
The speed matters, but so does the reliability. The tools that were fragile or disabled in nf-core/rnaseq now just work. No intermediate temporary files flooding disk, no R or Bioconductor dependency chain to manage. A 15-minute job fits well within spot instance lifetimes, which changes the economics of cloud execution. Users of the nf-core/rnaseq pipeline can now get preseq and TIN outputs for all samples.
RustQC produces near-identical output to the originals. Because of this, MultiQC picks up RustQC's output files as if the original tools created them, without any modifications. Deterministic tools like featureCounts produce byte-identical gene counts. This makes RustQC a drop-in replacement: swap it in, or swap it out, with zero risk to scientific conclusions. It accepts SAM, BAM, and CRAM input, handles gzip-compressed GTF files, auto-detects Ensembl and GENCODE biotype attribute formats, and validates the user-specified strandedness against the data itself, catching common configuration errors before it can affect downstream analysis. And of course it has a beautiful CLI with a dynamic progress bar and easy to read output.
You can find full benchmark data, reproduction scripts, and per-tool comparison tables in the RustQC documentation and the RustQC-benchmarks Nextflow pipeline.
Built with AI, Validated by Domain Expertise
The combination of domain expertise plus AI code generation is what made this feasible. Having reference outputs from the original R, Python, and C tools meant I didn't need to fully understand the Rust code, as long as I verified that the outputs matched what the original tools produced. Seqera AI sped this up massively - it could launch benchmark runs on Seqera, fetch task-level metrics from completed pipeline runs, and compare results, removing the friction from the validate-iterate cycle and allowing it to proceed autonomously. Because Seqera AI can even be used as a subagent of Claude and OpenCode, I could switch between different models for different tasks, all within the same interface. I’d describe what a tool should do, the LLM would generate Rust code, run the pipeline, compare outputs, I’d convince the LLM that yes, those differences do matter, then tell it to fix the mismatches, repeat.
I want to caution that there was significant work between "working beta" and something I'm happy to present to the wider world. Whilst the initial rewrite was done in a few days, weeks of validation and benchmarking followed - slowly inching towards output parity. This work is not glamorous and it would be easy to believe the LLMs assertions that the results are close enough. Stringency in this validation will be key to community adoption and trust. We've written more about the broader implications of AI-assisted rewrites and the principles we followed in our companion post and at rewrites.bio.
Nextflow: The Foundation for Agentic Bioinformatics
In order to function effectively, AI agents need fast, reliable, scriptable tooling - we’ve seen this recently with Anthropic acquiring Bun and OpenAI acquiring Astral. Nextflow is the bioinformatics equivalent of these tools. It is the orchestration layer where pipelines are defined, tools are composed, and results are validated. The same properties that have made Nextflow the standard for reproducible bioinformatics, such as modular processes, containerized execution and declarative configuration, make it the natural foundation for AI-driven bioinformatics. As AI agents take on more of the pipeline development and optimization work, a modular, containerised orchestration layer is what they'll build on.
Nextflow’s strength extends beyond testing individual tools. There is a huge ecosystem of Nextflow tool wrappers to start from, and as the rewrite progresses the finished sections can be solidified into a pipeline and with those steps being cached on reruns. The exact results can be locked using nf-test, ensuring no regressions. The end result can be a full benchmarking pipeline, which anyone can run on any infrastructure to replicate your results.
The modularity in Nextflow that made RustQC possible to build should hopefully also make it easy for the community to adopt. For anyone running nf-core/rnaseq, switching to RustQC is a single parameter change, replacing the QC subworkflow with a single RustQC process. Confidence in output equivalence is proved via nf-test snapshots, locking down the equivalence on process outputs.
Getting Started
RustQC is available now as a single compiled binary for Linux x86, Arm, and MacOS Intel / Apple Silicon. Install the binaries directly, grab them from Bioconda, or use the Docker image (see installation docs). Once you have the tool on your PATH, point it at one or more BAM / SAM / CRAM files and a GTF file and you're away:
All output files follow the naming conventions of the original tools, so existing MultiQC configurations and most pipeline configurations should work without modification.
The current scope is RNA-seq QC, as used by nf-core/rnaseq. Benchmarks have focused on human data (GRCh38), and RustQC implements the subset of each tool's features that nf-core/rnaseq uses. Other organisms, platforms, and features have not been formally validated yet, but it's my expectation that more tools and QC paths will be added over time.
Contributions, bug reports, and expanded validation on new organisms and data types are welcome. If you find a case where RustQC output differs from the upstream tool, open an issue with a reproducible example, and the validation suite will grow from there. Likewise, we'd love to hear what you think of rewrites.bio, my hope is that it starts a discussion within the community and helps us form consensus.
- →Documentation: seqeralabs.github.io/RustQC
- →GitHub: github.com/seqeralabs/RustQC
- →Benchmarks: github.com/seqeralabs/RustQC-benchmarks
- →Rewrite principles: rewrites.bio
Note: RustQC is open source and being released for community use. It is not an official Seqera product and is not officially supported by Seqera. We'd love to see what people think of it and whether it's useful for their work.