


We’re thrilled to announce the release of Nextflow 26.04, with new capabilities to help you write bug-free pipeline code and catch errors early. A new module registry and native nextflow module CLI commands make sharing and installing workflow modules simple.
Building on the foundation of workflow inputs/outputs and type annotations introduced in Nextflow 25.10, this release delivers several key features that make writing and maintaining high-quality bioinformatics pipelines easier than ever. Nextflow 26.04 introduces powerful new language constructs such as records and static typing, and takes a significant step forward in code reuse with the new Module Registry.
These features improve the experience of writing Nextflow code, whether you're doing it yourself or with an AI agent. Strict syntax and static typing produce specific, actionable error messages that a code-generation loop can parse and fix without human intervention. Records use named fields instead of positional indices, so generated code is less likely to silently break when inputs change. The module registry is fully CLI-driven: agents can search for modules, read their documentation, and run them directly, all through shell commands.
Strict Syntax
Preparing for strict syntax
In 2025 we released a completely new Nextflow syntax parser, which we call “strict syntax”. This parser introduced a stricter implementation of Nextflow DSL2, and laid the groundwork for new language features like static typing. Until now, it has been opt-in, but Nextflow 26.04 uses the strict syntax parser by default, bringing the rich error checking of nextflow lint to nextflow run. This also means that you can use new language features without needing to set NXF_SYNTAX_PARSER=v2 in your environment.
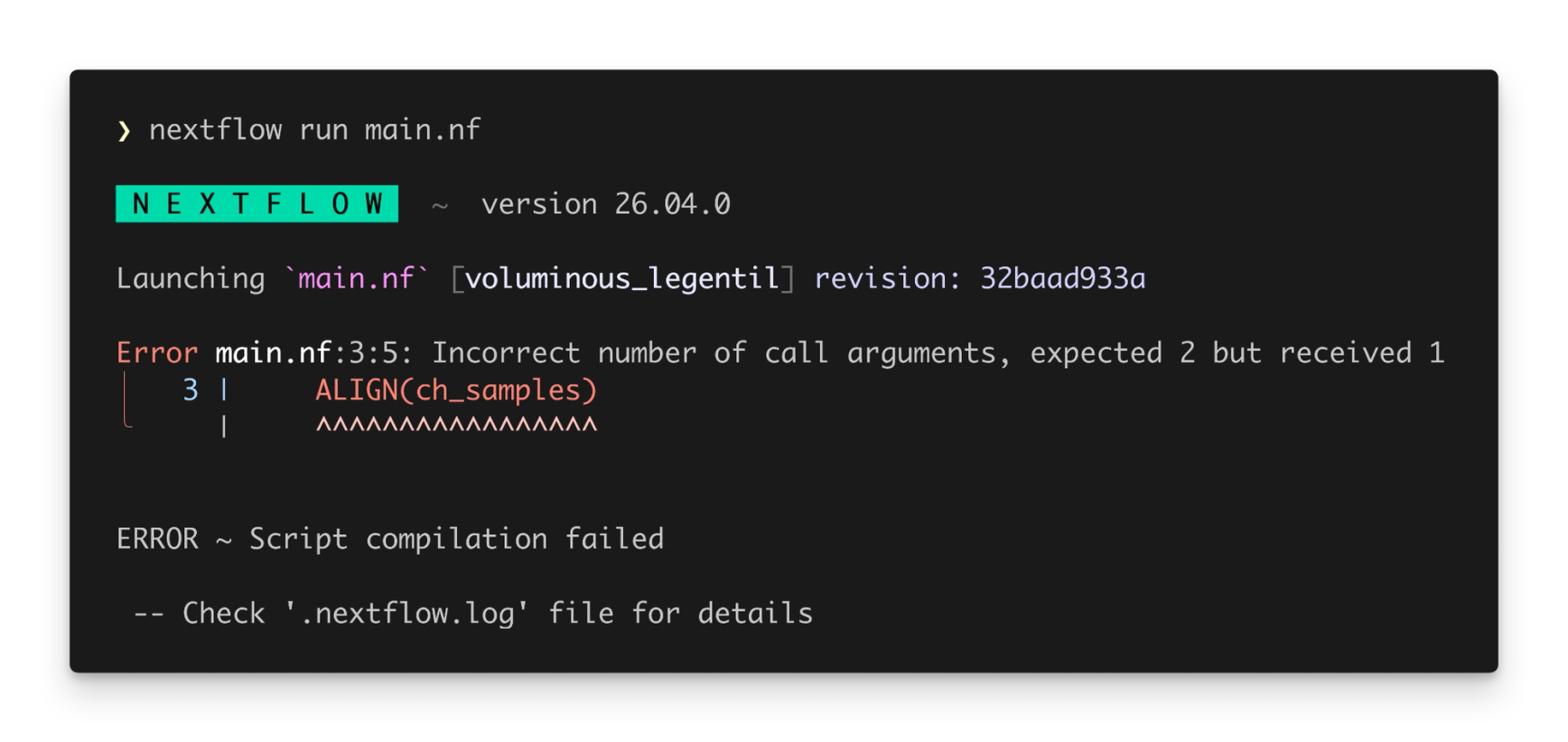
Some existing pipelines might not run out-of-the-box with Nextflow 26.04 – this can happen when a pipeline uses Groovy syntax that was not included in the Nextflow language specification. You can still run these pipelines by setting NXF_SYNTAX_PARSER=v1 in your environment. We recommend updating these pipelines to comply with strict syntax so that they can benefit from the improved developer experience and new language features.
Records and Record Types
Migrating to records
Nextflow 26.04 delivers on a long-awaited feature for the Nextflow language: records and record types. Most Nextflow pipelines have some form of structured data that is propagated through the pipeline. Multiple values need to be kept in sync so that Nextflow knows how to properly parallelise process execution. Records are a new way to structure this data as it flows through a pipeline. They are a replacement for tuples: where tuples contain elements that are accessed by index, records contain fields that are accessed by name.
To get started, create a variable with the new record keyword:
Since record fields use named keys instead of positional indices, their order no longer matters. This avoids a common pitfall in Nextflow pipelines where modifying a tuple input requires checking the order of arguments everywhere it’s called.
To document and validate records, you can also create custom Record types. These are named data structures that define the expected fields and types for a record.
If a record is supplied that does not match the expected record type, the Nextflow language server catches the error immediately:
💡 Note: Type checking is currently only performed by the language server. A future version of Nextflow will provide type checking innextflow lintandnextflow run.
Records in Nextflow aren’t like classes and objects in other languages:
- →Records are anonymous, which means that you can create them on-the-fly without an explicit type.
- →Record types are used to specify minimum requirements at the boundaries of a pipeline, workflow, or process. Any record that satisfies the requirements of a record type can be used, even if it has additional fields.
Records are designed to make it easy to model data at every layer of a pipeline, without bloating your pipeline code with type definitions and type conversions.
Static Typing (preview)
Migrating to static typing
Nextflow 25.10 introduced the first phase of static typing with type annotations and basic type checking. Nextflow 26.04 brings full support for static typing, with typed processes and typed workflows.
Processes
Typed processes now support record inputs and outputs:
As well as a streamlined syntax for tuple inputs, making it easier to adopt static typing before migrating to records:
Workflows
Typed workflows now provide first-class support for records and static typing in dataflow logic:
Several operators have also been updated for use with static typing and records. For example, the join operator can now join channels of records on a matching record field (such as id in the above example). See the best practices guide for more information about using operators with static typing.
Language server
The Nextflow language server can use all of this new information to validate the structure of your data at every step of a pipeline, including records. For example:
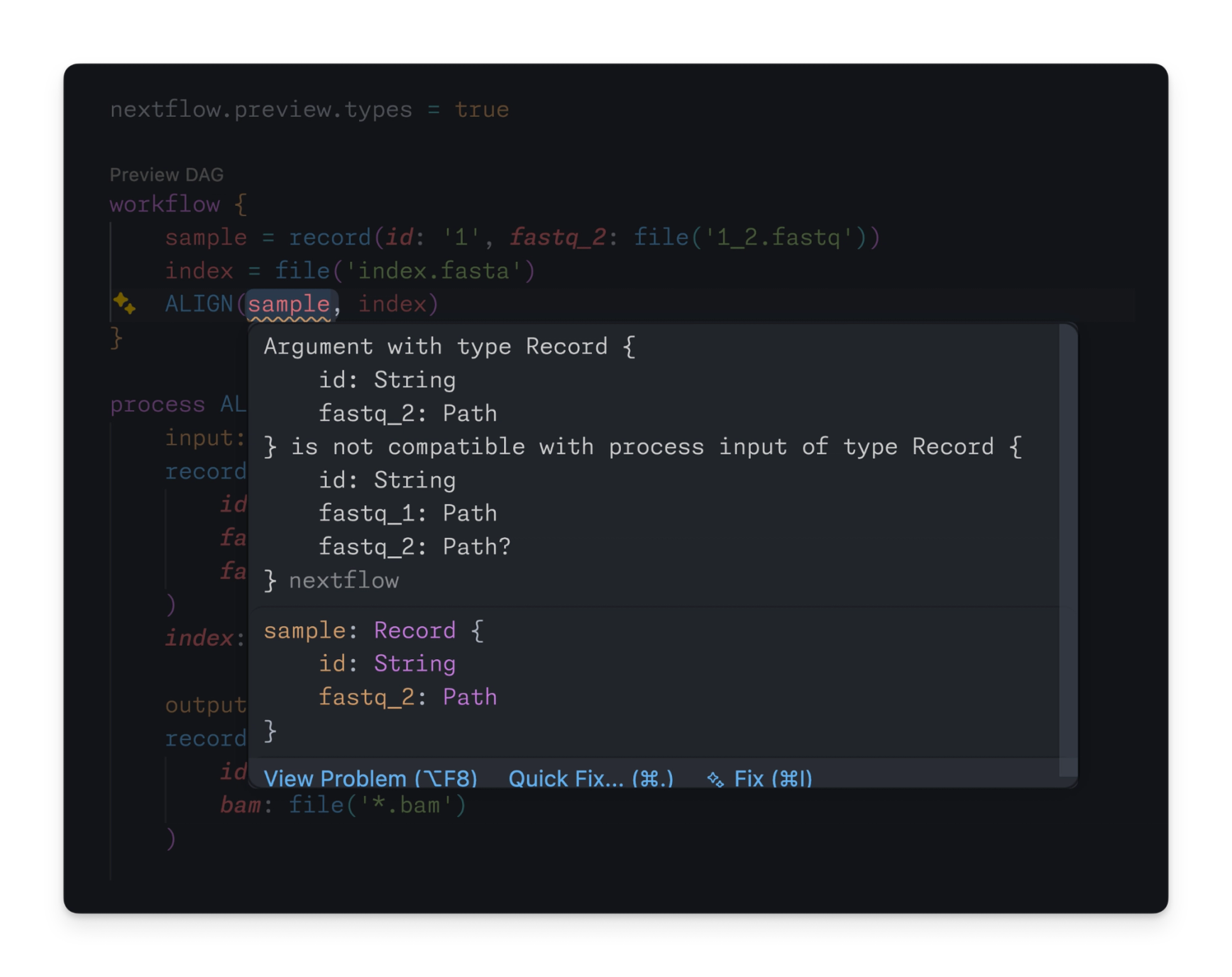
Looking ahead
Static typing (specifically, typed processes and typed workflows) must be enabled using the nextflow.enable.types feature flag. This is done separately for each script, allowing you to adopt static typing one file at a time. It remains in preview for Nextflow 26.04 and will become stable in Nextflow 26.10. We encourage everyone to experiment with static typing in their pipelines and share feedback, as we work towards stabilization.
Module Registry
Using registry modules
Nextflow 25.10 introduced the Nextflow registry for publishing and discovering plugins, which led to an explosion of new community plugins. With Nextflow 26.04, we have extended the registry to implement a native module system – a way to publish, install, and run modules through the Nextflow CLI.
Nextflow modules can now be published and shared through the Nextflow Registry, similar to plugins. You can use the nextflow module command to work with remote modules. For example:
Especially powerful is the nextflow module run command, which allows you to run a module directly without writing a pipeline:
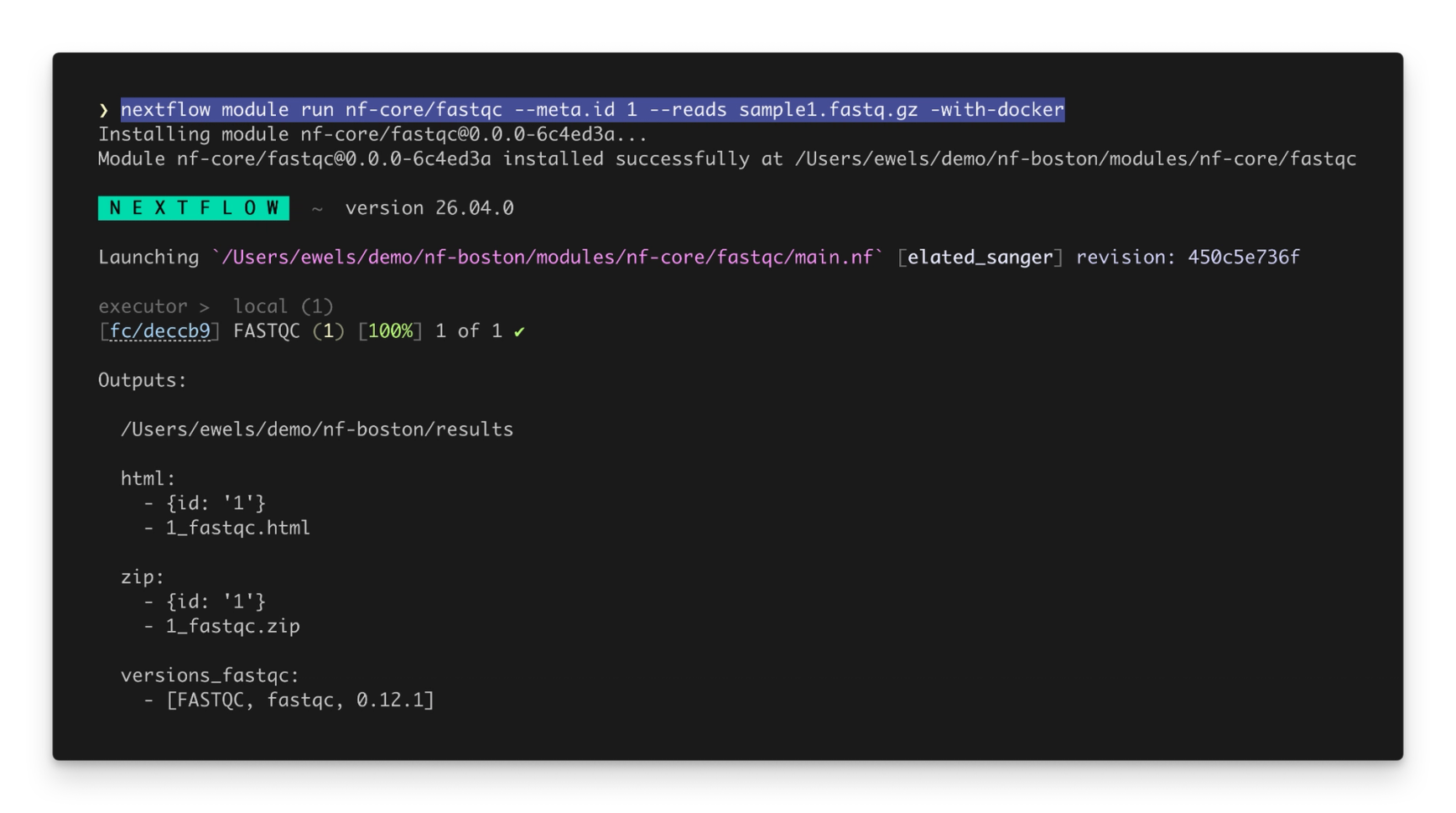
Process inputs can be specified as command-line arguments, and process outputs are published to an output directory and printed to standard output.
Modules are installed in the modules directory of a pipeline, following a standard community practice. As a bonus, you can include modules using their canonical name, saving you from the headache of relative paths:
All nf-core modules are automatically synced to the registry under the nf-core namespace, and can be used as nf-core/<name> as shown above. You can also publish your own modules by claiming a namespace in the module registry.
Get Involved
We hope that you’re as excited about Nextflow 26.04 and the new module registry as we are! As a community-driven project, we’d love for you to get involved:
- →Nextflow 26.04: Migrate to the latest release to access new capabilities with static typing and modules: Read the migration guide for Nextflow 26.04
- →Try out static typing: Let us know how static typing works for you and your pipelines. Learn how to migrate to static typing
- →Discover the Module Registry: Use the standard nf-core modules, or publish and share your own modules. Explore the Module Registry
The Nextflow ecosystem continues to mature with these updates, providing the foundation for more robust, maintainable, and enterprise-ready bioinformatics workflows. Whether you're writing pipelines by hand or letting AI agents generate them for you, Nextflow 26.04 gives you the tools to move faster and catch mistakes sooner.