
This is a joint blog post from Harshil Patel of Seqera, Rosi Bajari, and Bryan Lajoie of Element Biosciences describing a new integration between ElemBio Cloud and Seqera that provides hands-free, end-to-end automation for large-scale analysis workflows.
Automating NGS data analysis in the cloud is a significant challenge. Researchers need to deal with a wide variety of issues that include data management, setting up complex cloud infrastructure, running and monitoring pipelines, and organizing and interpreting results. Fortunately, Element Biosciences has made this much easier with a comprehensive integration between ElemBio™ Cloud and the Seqera Platform.
In this article, we introduce ElemBio Cloud, explain the challenges associated with running large-scale analysis pipelines, and describe a new integration between ElemBio Cloud and Seqera that provides hands-free, end-to-end automation.
About AVITI Sequencers
The AVITI™ system is a benchtop NGS device with dual independent flow cells, each capable of producing over 1 billion paired end reads per run with high accuracy. In a recent published benchmark, AVITI demonstrated consistent and accurate quality scores beyond Q401. The same tests showed how Element systems benefit the differentiated ability to sequencing libraries with insert sizes of 1,000 base pairs, resulting in increased variant calling comprehensiveness and accuracy.
Additionally, Element Biosciences provides exceptional affordability, delivering sequencing costs as low as $200 per genome or $2 per gigabase. This enables labs and research organizations to quickly produce high-quality data at a very low cost.2
Challenges with bioinformatics analysis
Before samples can be analyzed, basecall files generated by sequencers are typically converted into FASTQ format ⎯ a standard that encodes sequenced reads along with quality scores estimating the confidence of each base call. FASTQ files serve as the standard input to a variety of secondary analysis pipelines, including those from nf-core ⎯ a community effort to collect a curated set of analysis pipelines written in Nextflow. The AVITI system continually produces and streams “bases” files to the customers desired output location continually as the run progresses. Once the run completes, all bases files are ready to be transformed into the FASTQ file format by the bases2fastq software. In addition to building their own high-throughput bases2fastq-nf pipeline in Nextflow, Element has also contributed to the nf-core/demultiplex pipeline, which is used to demultiplex raw reads from AVITI and other sequencing platforms into FASTQ format.3
Even modest-sized studies can involve hundreds of samples, multiple instruments, and tens or even hundreds of terabytes of data. While some organizations have access to local HPC facilities with dedicated support teams, most organizations face significant challenges collecting and analyzing data at scale. Some common challenges include:
- →Retrieving, organizing, and managing terabytes of data – locally or in the cloud.
- →Generating FASTQ files from raw reads.
- →Performing secondary and tertiary analysis on samples and organizing results.
- →Provisioning and managing cloud computing environments.
- →Ensuring that analysis is reproducible and scalable for larger cohorts.
- →Managing and monitoring cloud-related costs to avoid overspending.
These issues are challenging for large organizations, but are even more daunting for small research teams with limited staff. Researchers need a solution to manage this complexity while allowing them to run scalable data analyses in their preferred cloud(s).
Automating analysis
When performing analysis at scale, automation is essential. It’s often impractical for most technicians to load samples, manage computing environments, manually move results to cloud storage, trigger pipelines for FASTQ generation and downstream secondary analysis. This is not only time-consuming and labor-intensive, but these types of tedious processes inevitably result in errors. Ideally, technicians should be able to focus on loading and sequencing samples, with all these additional steps handled automatically behind the scenes.
The idea of automating end-to-end analysis with AVITI sequencers is not new. In 2022, Element and Seqera collaborated on a technical tutorial explaining how to leverage AWS Lambda, Seqera Datasets, and the Seqera CLI to automatically process basecall files streamed to cloud storage. This solution is described in the article Workflow automation for Nextflow pipelines. While this is a good solution, it has a few limitations:
- →The solution relies on services such as AWS Lambda, AWS Secrets Manager, and AWS CloudWatch and is thus tightly coupled to the AWS ecosystem.
- →It requires a significant amount of initial setup by people skilled at deploying and configuring cloud infrastructure in AWS.
- →The solution doesn’t address monitoring, so if something goes wrong it can be difficult for users unfamiliar with how the solution works to troubleshoot problems.
ElemBio Cloud and Seqera
Fortunately, the Element Bio team has been hard at work and recently announced a more robust integration with the Seqera Platform. The new integration is implemented as a feature of ElemBio Cloud, an online platform used to manage AVITI instruments, monitoring runs, visualizing data, and managing team members. A high-level illustration of the workflow is provided below.
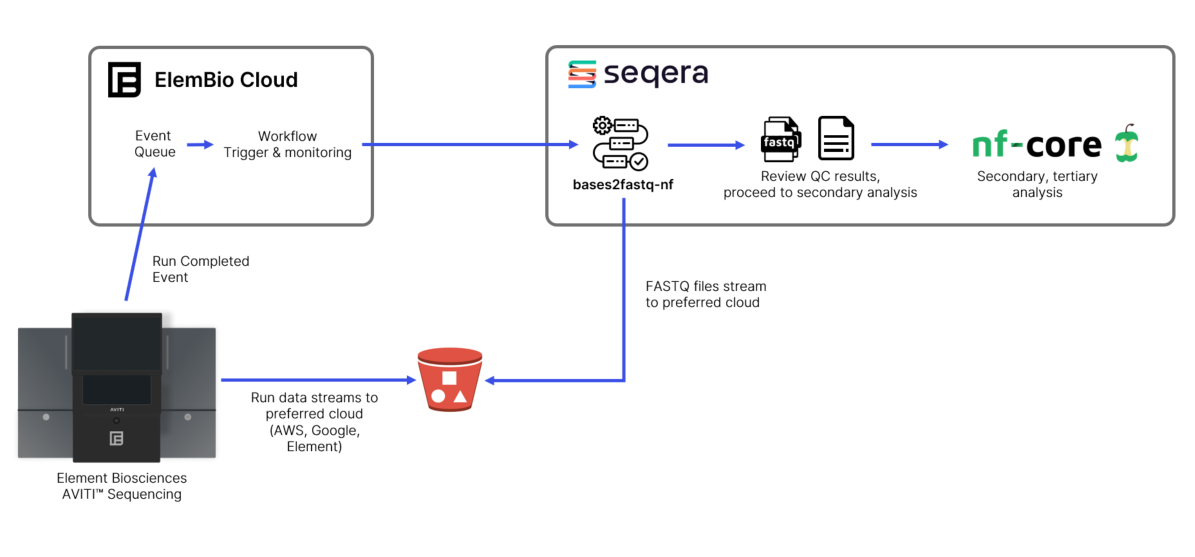
To enable the integration, AVITI users simply log into ElemBio Cloud and configure details about their storage and Seqera environments using the portal’s interface. Users can manage multiple AVITI sequencers from their ElemBio Cloud account and provide cloud storage credentials enabling sequencer(s) to stream analysis results in a standard format to their preferred cloud object store.
ElemBio Cloud users can easily configure the Seqera Platform as a compute provider by entering an Access Token retrieved from the Seqera Platform web UI as well as providing the Workspace ID and Organization ID where Element’s bases2fastq-nf pipeline will run. The setup and management of the user’s preferred cloud infrastructure is fully automated by Seqera. Once the storage and Seqera compute integrations have been set up, the user can set up an ElemBio Cloud flow, configuring the launch and trigger details, including the ability to automatically launch a demultiplexing workflow as soon as a run completes.
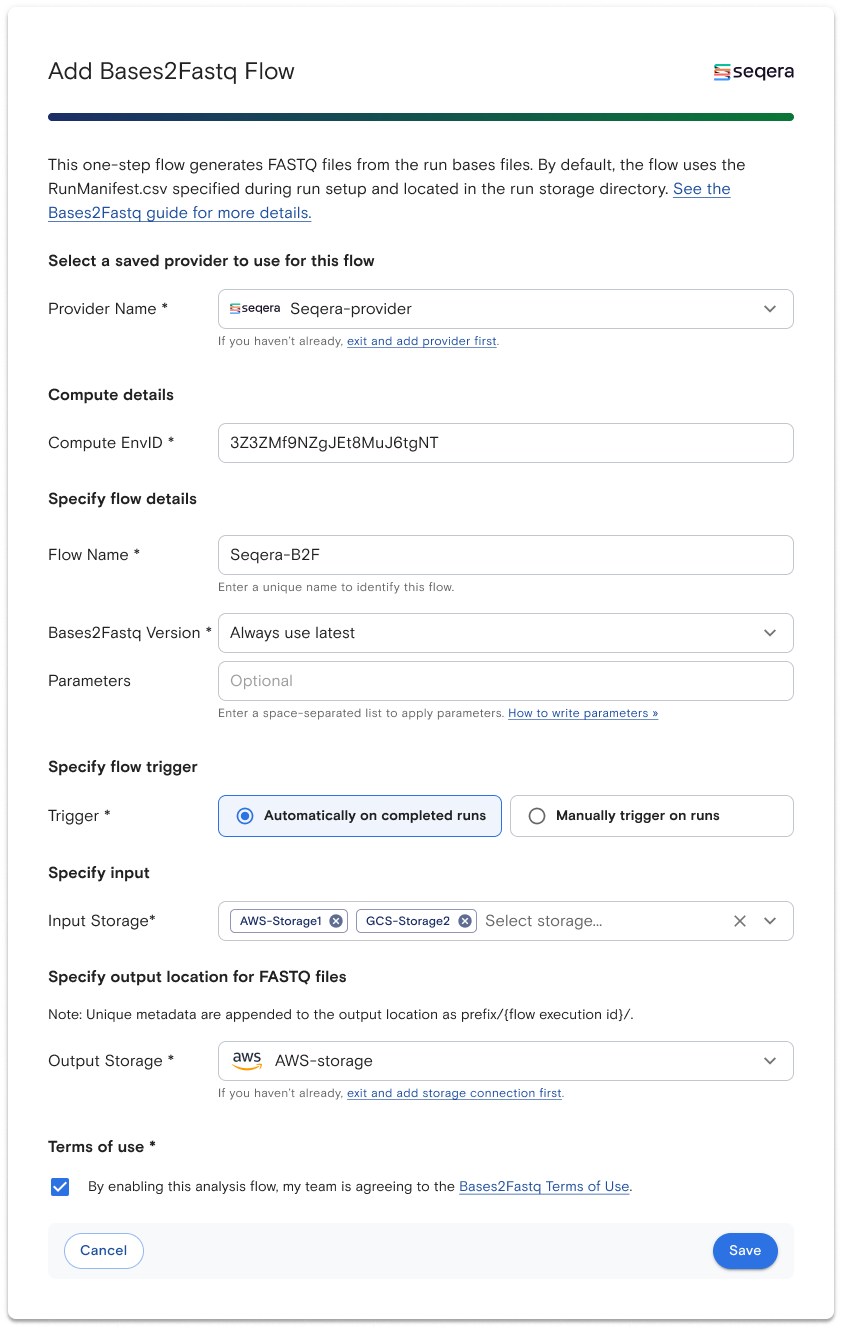
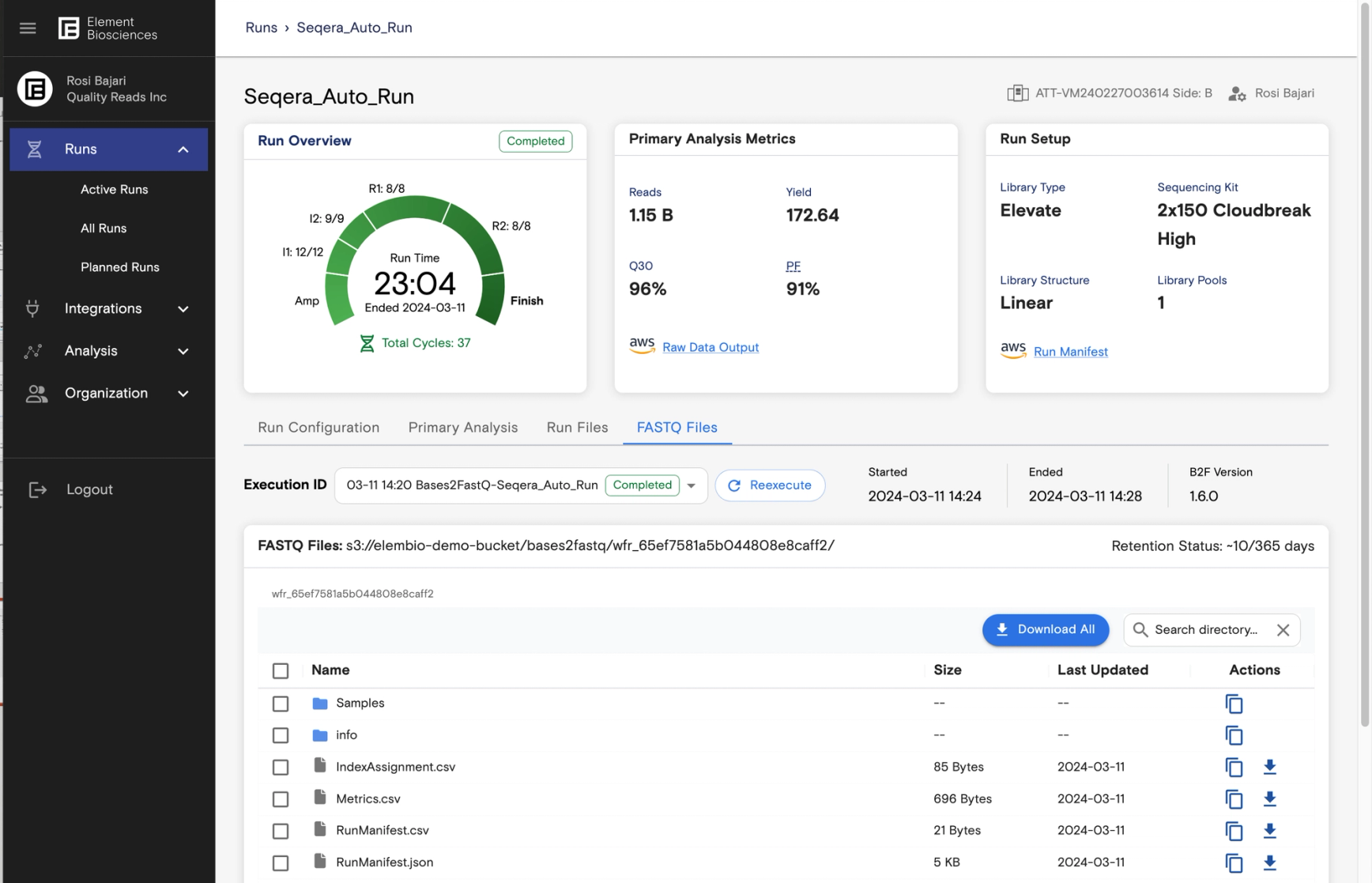
As the sequencer runs, data is automatically streamed to the designated object store. On completion, the AVITI sequencer automatically triggers a workflow in ElemBio Cloud that, in turn, securely calls the Seqera Platform to optionally provision cloud resources in the user’s cloud of choice The Seqera Platform then retrieves and runs Element’s optimized bases2fastq-nf pipeline stored in the Element GitHub repo, performing the sample demultiplexing and FASTQ conversion. After triggering, ElemBio Cloud continues to monitor the pipeline and upon completion allows resulting data to be browsed, downloaded, and shared. Element Biosciences cannot access any sequencing data transferred to connected accounts.
Using the ElemBio Cloud interface, AVITI users can provide details such as the version of the pipeline to run, specify how pipeline runs are triggered, and provide separate input and output storage buckets.
This integration provides several advantages over earlier efforts at achieving end-to-end automation:
- →It is easy to configure and use, requiring minimal expertise on the part of AVITI platform users.
- →ElemBio Cloud users can stream data to their preferred cloud storage environment, including Amazon S3 and Google Cloud Storage.
- →Users don’t need to concern themselves with provisioning cloud infrastructure or launching pipelines — this is handled transparently by the Seqera Platform.
- →Users can monitor pipeline runs in real-time either via the ElemBio Cloud or Seqera Platform interfaces with extensive reporting and visibility to runtime costs.
- →Researchers can easily perform secondary and tertiary analysis, optionally leveraging nf-core pipelines to run additional downstream analysis directly from the cloud object stores containing the collected FASTQ files and QC data.
In addition to providing convenience and improving efficiency, using the Seqera Platform with AVITI to manage pipeline execution can significantly reduce costs. Seqera Platform resource optimization can help right-size task resource requests based on historical resource usage to avoid costly over-provisioning. In production scale pipelines, Seqera’s resource optimization solution has been shown to improve throughput by up to ~40% and reduce cloud-related compute costs by up to 85%!4 Additional optimizations are available by using Seqera’s Fusion file system.
Looking forward
The ElemBio Cloud – Seqera Platform integration was recently announced at the Nextflow Summit in Boston and in a joint Element-Seqera webinar titled AVITI™ Analysis at any Scale.
Element has outlined an exciting roadmap for ElemBio Cloud with new features, including expanded support for secondary analysis, file browsing with connected accounts, and enhanced tracking and visualization of orchestrated workflows. Element is also adding bases2fastq support to Seqera’s MultiQC reporting tool, exposing key metrics and making it easier for ElemBio Cloud and Seqera Platform users to view and analyze QC metrics in one place.
Access to the integration became available in March 2024, and AVITI users can contact Element support for more information about participating in Element’s programs.
---
1 Accurate human genome analysis with Element Avidity sequencing – biorvix.org - August 2023
2 Element delivery $200 Genome on AVITI™ Benchtop Sequencing System – January 2023
3 While nf-core/demultiplex is an excellent general-purpose solution, Element Bio also has a separate pipeline optimized for AVITI for Element Biosciences customers
4 See Optimizing resource usage with Nextflow Tower for details. This example is based on the production-scale nf-core/viralrecon pipeline. Before optimization, pipeline costs were USD 9.97 using AWS spot instances for a single run. After generating and applying optimized resource recommendations in Seqera, the same pipeline run cost just USD 1.54, an ~85% reduction.