

Building a Scalable Protein Design Pipeline with Seqera AI for Adaptyv's Nipah Binder Competition
💡Note: This guide shows you how to effectively use AI to build reproducible pipelines to design proteins. In this tutorial we use three key pieces of technology:
• Seqera AI
• Nextflow
• Seqera Cloud + Kubernetes
These technologies combined, have allowed us to build a pipeline that designed novel binding proteins for the Nipah Binder competition on Proteinbase. All you need to replicate this workflow is:
• GitHub account
• Access to Seqera Cloud
• Nebius AI cloud account with credits
At Seqera, we’re dedicated to making scientific data analysis accessible at scale, supporting teams across diverse areas such as genomics, imaging, protein analysis and drug discovery. Although we don't focus on creating models for protein design or drug discovery ourselves, we recognize the remarkable technological advancements and successes in this field. Here, we demonstrate how we used Seqera AI to build a scalable protein design pipeline that helped us rank 6th (based on ipSAE ranking) in Adaptyv’s Nipah Binder Competition on ipSAE prediction.
In 2024, we developed a modest, example Nextflow pipeline for protein design to present at the Nextflow Summit in Barcelona. This initial effort was time-consuming and required significant manual work, as we had to build every component from the ground up. Simply getting a single container to build correctly was difficult. Connecting different tools demanded in-depth knowledge of each tool's specific requirements and syntax. Furthermore, the results from design tools at the time, while promising, frequently resulted in designs with low binding efficiency.
Since then, the landscape has undergone a fundamental transformation. While the underlying biology remains complex, AI-based tools have matured to a point where they genuinely and dramatically speed up pipeline development. What once took weeks of difficult iteration can now be accomplished in days (or even hours), yielding comparable or superior results. Concurrently, the tools available for drug discovery and protein design have advanced so much that even individuals new to the field can generate high-quality binding candidates for discovery campaigns.
This blog post details the creation and deployment of a proof-of-concept Nextflow protein design pipeline, nf-proteindesign. Specifically, we cover:
- →Designing the Nextflow pipeline from scratch using Seqera AI
- →Testing and deploying the pipeline on Seqera utilizing Nebius managed Kubernetes.
- →The pipeline's application in the Adaptyv Nipah Binder Competition, where it successfully helped us design protein binders and achieve a 6th place ranking on the leaderboard.
The Adaptyv Nipah Binder Competition
The second protein binder design competition - hosted by Adaptyv - is a fantastic event that brings together experts and enthusiasts in the protein design field to try and create novel binding proteins against important targets for human disease. While the 2024 competition focused on EGFR (an important target in fighting cancer), the 2025 competition focused on designing binding proteins against Glycoprotein G of Nipah virus. This zoonotic virus with an extremely high mortality rate (up to 75%) can jump from bats to humans - sometimes via other animals. Named in the WHO R&D Blueprint list of epidemic threats, identifying novel treatments or vaccines against Nipah virus is critical to combat this dangerous pathogen.
Envisioning a Protein Design Pipeline
💡Note: Nextflow is the leading open-source workflow orchestrator that simplifies writing and deploying compute and data-intensive pipelines at scale on any infrastructure. With Nextflow, you can easily create, share and manage reliable, reproducible scientific workflows that are portable across compute environments.Learn more about Nextflow
To begin developing our protein design Nextflow pipeline, we first determined the necessary analysis steps and corresponding tools required. Although we considered starting with the automatic identification of protein binding sites (hotspots) on the target protein, several excellent tools are available online for this task (Pesto, ScanNet,SPPIDER).
For the Nipah Binding Competition, we utilized Pesto to locate potential binding sites on the Glycoprotein G structure. Our process involved taking the sequence of the extracellular domain from Proteinbase (residues 71–602 of Glycoprotein G), predicting its structure using Boltz2, and then inputting the resulting pdb structure into Pesto. Pesto successfully predicted high-confidence binding interfaces in two regions: the N-terminal stalk region and the well-known EphrinB2 binding region.
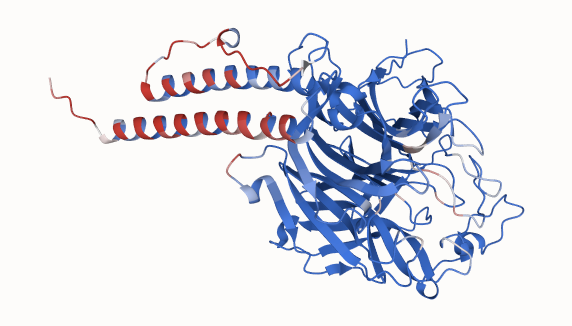
Figure 1. Pesto protein binding interface prediction for Nipah virus Glycoprotein G extracellular domain from the Adaptyv Protein Binding Competition. High confidence predicted binding residues are shown in red, residues with a low binding prediction score in blue.
We selected Boltzgen, a recently released tool based on the Boltz architecture, for our protein design. Boltzgen has demonstrated highly promising results in generating high-quality binding candidates. Although Boltzgen provides a ranked list of candidate binders on its own, we aimed to integrate additional downstream tools to improve our designs. Nextflow is perfectly suited for this, allowing us to easily link together containerized applications written in various languages and requiring diverse hardware (such as GPUs and CPUs). Our first step was sketching out a basic workflow using open-source tools, focusing on the desired functionality without immediate concern for the specific language or implementation details (Figure 1).
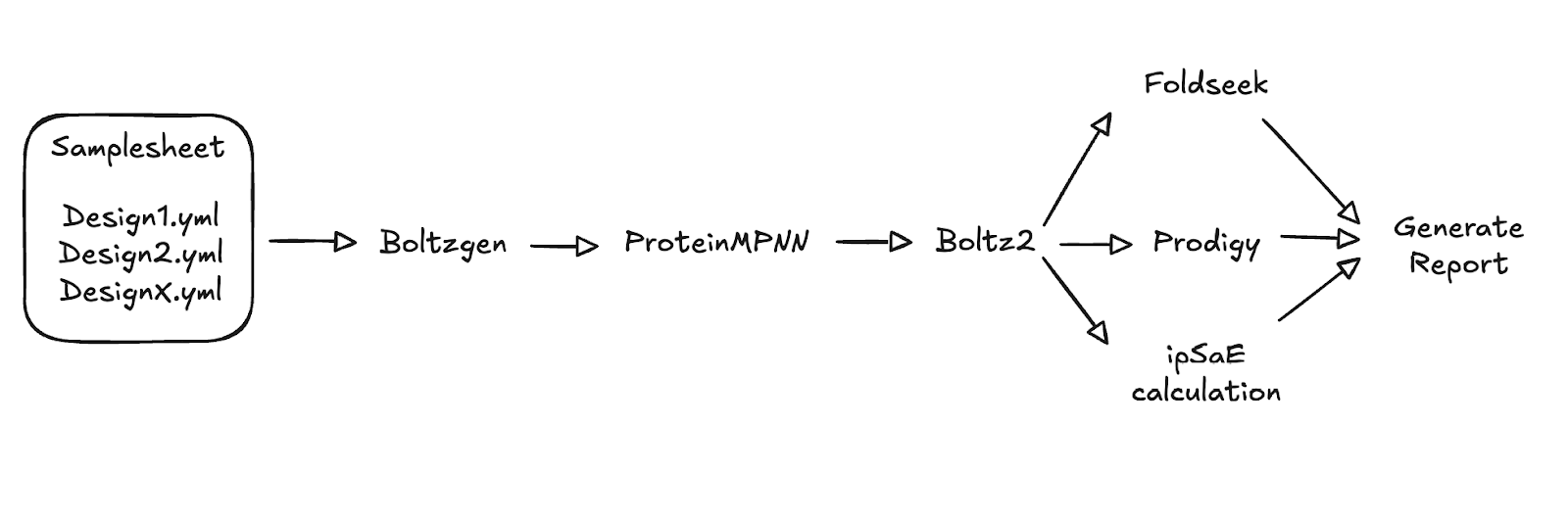
Figure 2. Sketch of the envisioned Nextflow pipeline for designing novel protein binders using a collection of tools.
Creating a Nextflow Workflow with Seqera AI
💡Note: Seqera AI is the bioinformatics agent built for the R&D lifecycle. Seqera AI enhances research productivity by answering complex questions, generating entire bioinformatics pipelines, validating code, and interpreting scientific results with high accuracy.Try Seqera AI now
After sketching out our envisioned pipeline, it was time to actually write the code. Seqera AI offers an asynchronous flow that allows it to generate commits and pull requests directly on a dedicated github repository. We started with a completely blank page (GitHub repo), connected it to Seqera AI, and described the protein flow we wanted to build, step-by-step.
We began by describing the pipeline architecture we had envisioned, starting with the first process Boltzgen. Within minutes of describing this flow, Seqera AI had generated an initial commit with a Nextflow process for BoltzGen. The code wasn't perfect - the container image Seqera AI had found was not actually for Boltzgen but for a different tool with the name boltz for example. Finding working container images on the internet is a general challenge for LLMs. Seqera is working to improve this by providing container image builds using our container provisioning service Wave for such cases. While the code wasn’t fully functional on the first try, using Seqera AI brought us to 90-95% of what we needed in a fraction of the time it would have taken us to write it by hand.

Figure 3. Commit message from Seqera AI implementing initial Nextflow pipeline using Boltzgen for protein design.
Seqera AI, used directly through our browser, was instrumental in developing the nf-proteindesign pipeline. We iteratively described the desired workflow components via chats with Seqera AI, which generated the entire pipeline over the course of 108 commits and 30 Pull Requests. The final product is a complete, end-to-end pipeline that combines various protein design tools with custom scripts. Thanks to Nextflow, this pipeline is containerized for reproducibility and can be deployed on any infrastructure, freeing us from dependence on a specific cloud provider.
Nextflow Pipeline Testing on Nebius AI Cloud
Now that we have an end-to-end Nextflow pipeline for designing protein binders, we had to test it and make it fully functional and performant on GPU infrastructure. As mentioned, while AI generated code gets us the majority of the way, human intervention is required to fix some of the current limitations of AI agents, such as assigning wrong or non-existent docker images to processes and sometimes using the wrong output from a tool.
💡Note: AI agent sandboxes are evolving quickly to be able to test written code on diverse infrastructure and correct their mistakes themselves but at the time of developing this pipeline, this still required some manual interaction with the code for testing.
To test our pipeline on GPU accelerated infrastructure, we partnered with Nebius to test our protein design pipeline on their AI Cloud. Nebius provides inexpensive, on-demand access to powerful GPUs such as H200, H100 and L40s via virtual machines, Kubernetes clusters or Slurm in Kubernetes (Soperator). We have previously covered how to use Soperator with Seqera using the Slurm executor. For this blog post, we ran Nextflow using GPU virtual machines and the managed Kubernetes service from Nebius with Seqera.
💡Note: Seqera is the bioinformatics platform that unifies pipelines, data, interactive analysis, AI, and compute across the scientific data lifecycle. Built by the creators of Nextflow, Seqera enables scalable pipeline orchestration with complete traceability and real-time monitoring.Discover Seqera Platform
First, to quickly test and make our pipeline functional, we used a virtual machine with a single H100 GPU and directly connected to it via ssh. We had Seqera AI create a test profile for our pipeline that used parameter settings that allowed us to run the pipeline end-to-end in a few minutes. During testing, we identified minor functionality issues such as non-existing docker images or parsing of wrong output from specific tools. These are still some of the remaining limitations of LLMs and agents for code generation that will get better as foundation models improve, as we tune Seqera AI and we connect more of our tooling such as Wave to actually generate functional containers rather than discover them online. Thanks to Nextflow’s resume feature, we could fix each process step by step without having to rerun the entire pipeline each time. The result was a fully functioning pipeline run with GPU acceleration on the Nebius VM. Now that our pipeline was fully functional, we were ready to submit our production pipeline run to generate some real candidate binders for Nipah virus Glycoprotein G.
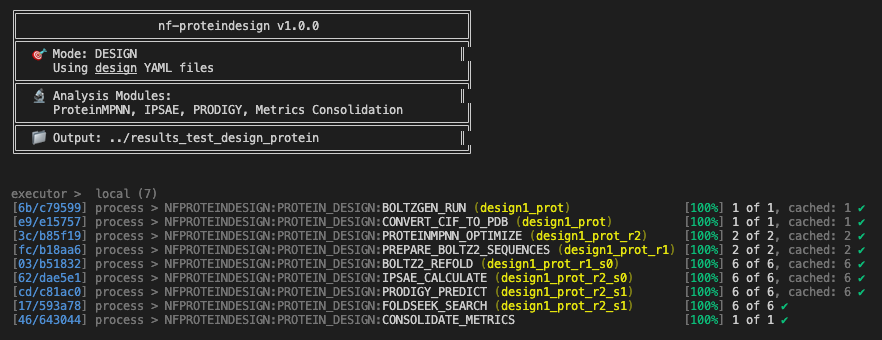
Figure 4. Nf-proteindesign Nextflow pipeline running end-to-end on a Nebius VM with an H100 Nvidia GPU.
Scaling Protein Design using Kubernetes, Nextflow and Seqera
To run our pipeline at a larger scale using multiple Boltzgen designs followed by redesigning sequences via ProteinMPNN, refolding via Boltz2 and downstream analysis, we deployed nf-proteindesign on a Nebius managed Kubernetes cluster. Nebius provides hosting of Kubernetes clusters without any nodes at zero cost, allowing intricate setups to fine tune your k8s cluster. Thanks to the Nextflow k8s executor, a Nebius Kubernetes cluster can be easily connected to Seqera for deployment of Nextflow pipeline runs.
The final run of our nf-proteindesign pipeline focused on designing binders against the Nipah virus Glycoprotein G. We used Boltzgen with four distinct design templates to create binders ranging from 50 to 250 amino acids in length:
- Unconstrained: No constraints were applied, allowing Boltzgen to target any region of the protein.
- Targeted Region 1: Specifically targeted residues 1 to 94.
- Targeted Region 2: Targeted specific residues: 235, 255, 256, and 281.
- Targeted Region 3: Targeted residues 332, 371, 388, 434–438, 489, and 490.
These specific target residues for the constrained templates were derived from binding site predictions made by Pesto. We performed 500 initial designs with Boltzgen, selecting the top 10 for subsequent redesign. Using ProteinMPNN, we generated 4 new sequences for each of the top 10 designs, resulting in a total of 200 final designs from the entire pipeline (40 initial Boltzgen, plus 160 ProteinMPNN redesigns).
A key aspect of the analysis phase was the scaling of our Kubernetes (k8s) cluster. We dynamically adjusted the cluster size from 8 to 24 H100 GPUs to match the infrastructure demands—specifically the number of parallel tasks—at different stages of the workflow. This flexible, autoscaling approach to Kubernetes infrastructure ensures both cost-effectiveness and scalability for production workloads.
Final Protein Design Structure
With our ranked candidate binders at hand, it was time to submit to the competition. As other participants have reported, the ipSAE scores calculated by the competition often differed from estimations, due to a combination of factors, including randomness in the co-folding predictions by Boltz2. As the competition considered the average ipSAE score of all submitted sequences for ranking, we decided to only put our best guess forward and chose the sequence with the highest ipSAE based on our calculations of our 200 candidates. Our top binder (nipah_design2_r003_s1) had a predicted ipSAE of 0.7896 in our predictions and scored even higher (ipSAE of 0.858) in the final competition. This binder was the third rank in the Boltzgen design (r003) and is a redesign with ProteinMPNN (sequence 1 out of 4) targeting the N-terminal stalk region of the Glycoprotein G.
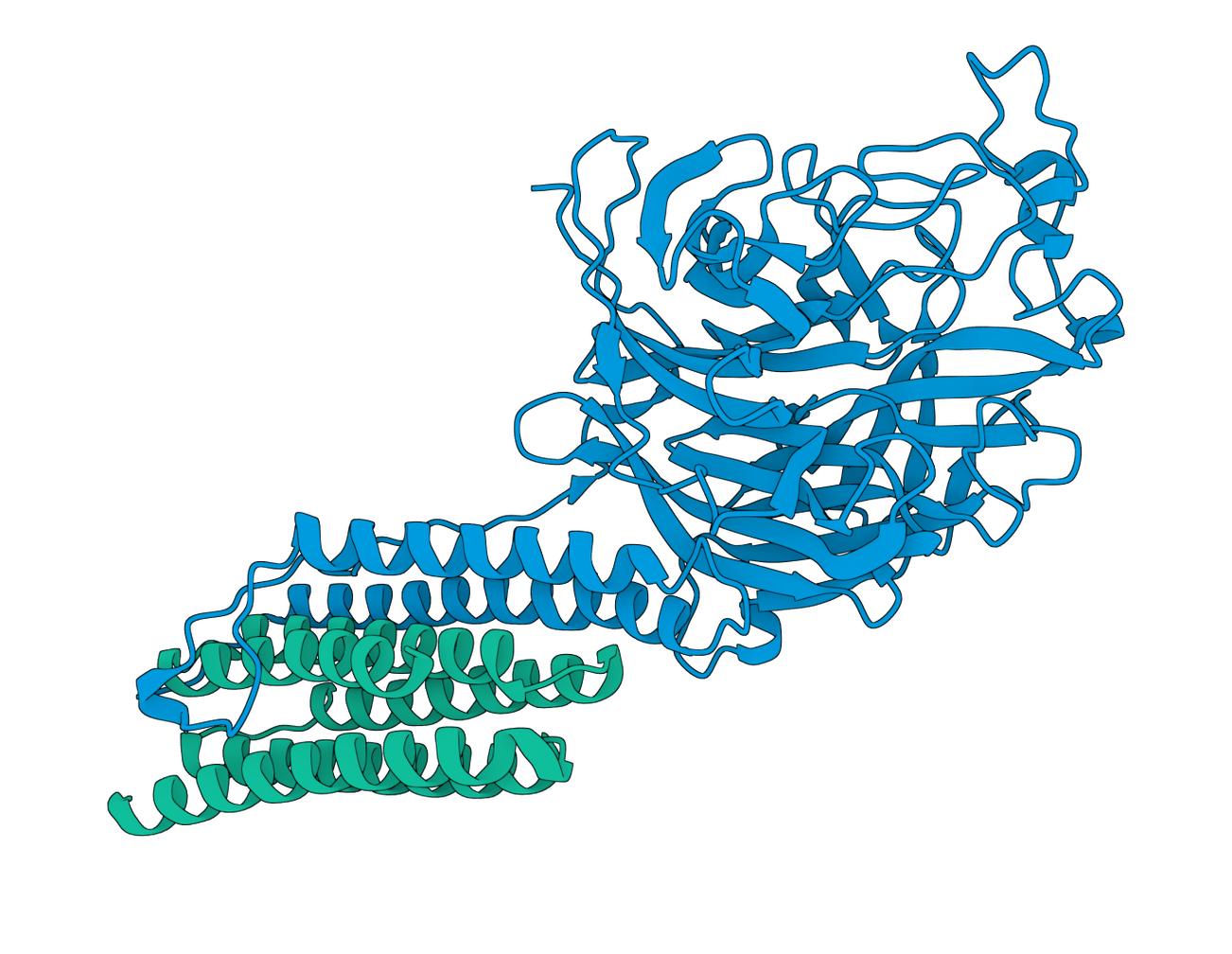
Figure 5. Folding prediction of Seqera’s best designed binder protein (nipah_design2_r003_s1) against Nipah Virus Glycoprotein G extracellular membrane sequence.
While we were somewhat lucky that the ipSAE estimation from the competition predicted the ipSAE of our binder favourably (even better than our estimated ipSAE) to rank 6th in this competition, we believe the competition in general highlights some key developments in the field:
- Protein design methods have reached a tipping point where anyone can design proteins at scale
- Orchestration of different methods using tools like Nextflow and Seqera, combining open source and potentially in-house protein design tools across any infrastructure provides incredible power and flexibility
Nipah Binder Competition - Wet Lab Validation and Key Takeaways
The wet-lab validation results are in, and unfortunately, our candidate binder did not bind to Nipah Virus Glycoprotein G. While it is, of course, disappointing that our design didn’t bind, it highlights the importance of testing actual binding of designs experimentally. The overall results of the competition were extremely exciting with 1030 proteins validated with an expression rate of 86% and 103 designs that showed binding to Nipah Virus Glycoprotein G. Unsurprisingly, the top hits based on binding affinity (KD) were not de novo designs, with the two top binders being redesigns of EphrinB2, the host cell receptor target of Nipah Virus Glycoprotein G.
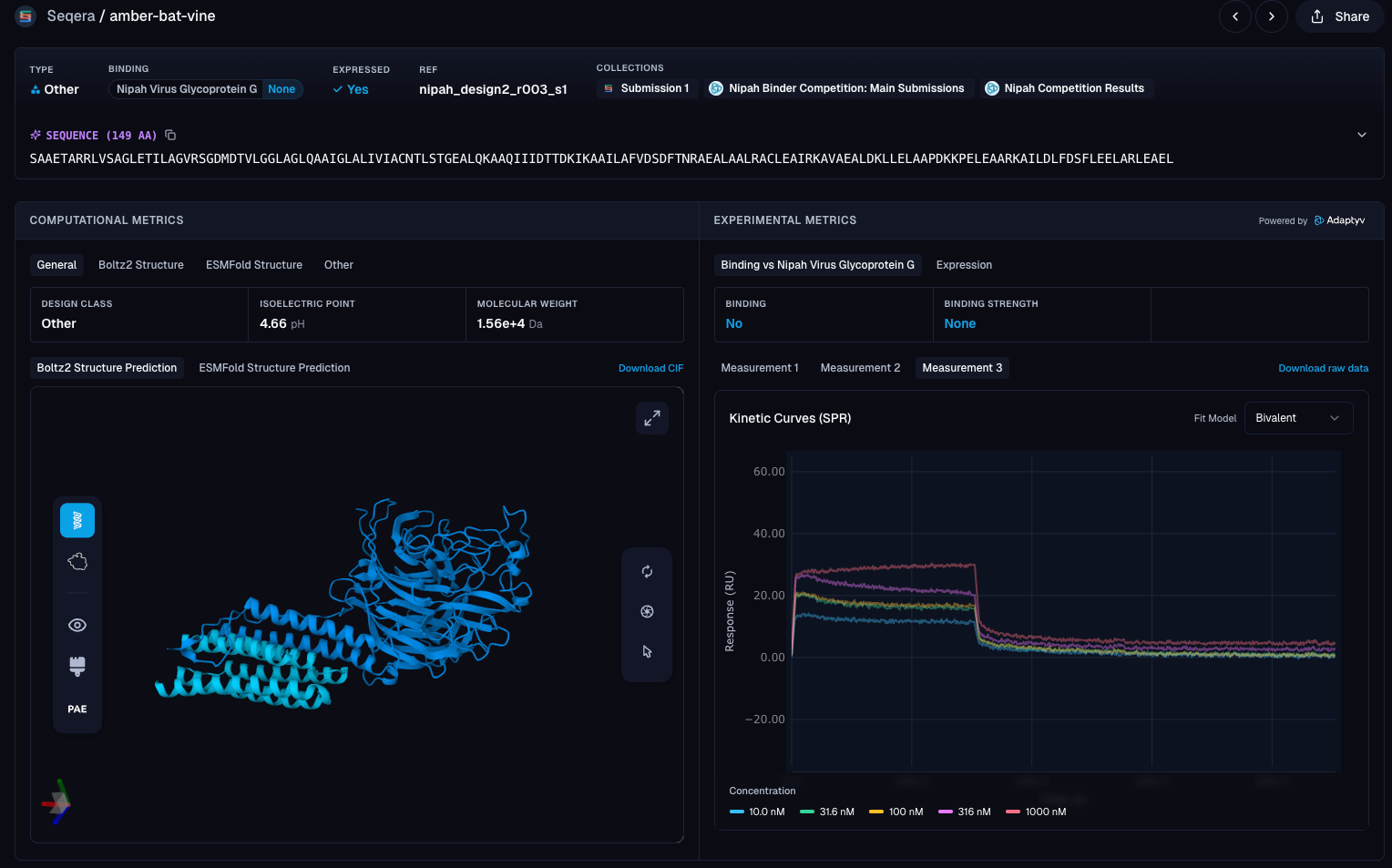
Figure 6. Computational and experimental results for Seqera's entry into the Nipah Binder Competition, as calculated by the competition hosts.
A number of de novo designs however, also showed strong binding, most notably designs by Nick Boyd and Sam Guns (Escalante Bio), which also ranked highly based on ipSAE in the leaderboard. They outline their reductionist approach to design binders and discuss why their designs performed so well in the competition in this blog post. Interestingly, all of their strong binders also target the stalk region of the Glycoprotein G, similar to our design, suggesting that it is indeed possible to design high-affinity binders against this region (at least in the experimental setup used by Adaptyv).
So why did our design targeting the stalk region not bind? Both Escalante’s designs and ours are made up of alpha helical bundles targeting the stalk alpha helix. We might have limited Boltzgen's design space by constraining our target epitope to residues 1-94 of the Glycoprotein stalk region. This suggests (at least for this particular target) that using tools like Pesto to identify good binding interfaces worked well, but restricting the design tool (Boltzgen) to only target those residues might limit design exploration.
The Nipah Binder competition results overall highlight the rapid pace at which this field is moving, with new methods for designing binders and antibodies being released frequently (the competition shows 97 different design methods used in total). We are excited to see how this field evolves and believe Nextflow and Seqera can help augment existing and new design workflows to make them scalable and reproducible on any infrastructure.
Conclusion
This project demonstrates the transformative potential of combining AI-powered pipeline development with scalable cloud infrastructure for computational biology. Using Seqera AI for iterative code generation across 100+ commits, we transformed weeks of manual pipeline construction into days of guided development. The resulting nf-proteindesign pipeline seamlessly orchestrates multiple specialized tools while maintaining reproducibility through containerization. Most importantly, this approach democratizes bioinformatics, allowing researchers to rapidly prototype and deploy scalable analysis without deep infrastructure expertise.