
Bioinformatics tools and workflows are rapidly transforming to address increasing complexity and demands for reproducibility, scalability, and collaboration. At the Nextflow Summit last month, I presented a retrospective on this evolution in the industry—here's my view of how the work of a bioinformatician has changed over this time span.
1. The Dark Ages: Manual Execution or Bash Scripts
In the early days, bioinformatics was overwhelmed by complexity and manual inefficiencies. Researchers relied on either fully manual execution or handcrafted Bash scripts using locally installed software versions. For example, a whole-exome variant calling analysis may start relatively simple, with a command to trim some sequencing reads using fastq:
Figure 1. The initial Bash command for preparing sequencing reads.
But very rapidly grows in complexity as the analysis spans alignment, different variant calling and variant annotation tools.
Figure 2. Completing WGS Analysis requires many more such Bash commands.
By the end of your analysis, you’ll find you might have used as many as 15 different software tools across up to 100+ individual command executions. And at every step of the way, you’re dealing with challenges in tracking files, software versions, and more.
Key Challenges
- →Traceability - no git versioning, limited logging
- →Reproducibility - no software containers, “it runs on my machine"
- →Scalability - reliance on hard-coded paths and infrastructure
The combination of poor reproducibility, scaling difficulties, and the constant need for manual intervention created significant barriers to scientific progress. These problems were overwhelming the genomics field, setting the stage for modern workflow management systems like Nextflow.
2. The Industrial Age: Nextflow
Discover Nextflow
Nextflow represented a significant shift in bioinformatics through the adoption of modern software tooling. Nextflow provided an integrated solution to the “dark ages” of Bash wizardry by introducing containerization for reproducibility, standardized reusable workflow definitions, and sophisticated resuming and error handling capabilities for incremental execution. Instead of executing all of the command executions manually, Nextflow provides a single unified CLI that could run analyses from beginning to end with traceability, portability, and scalability throughout.
Figure 3. An industrial WGS analysis completed with one command execution using Nextflow.
Despite a vastly improved experience for individual researchers, there were still many challenges encountered by teams trying to collaborate on their computational analyses:
Key Challenges
- →Visibility - Lost datasets, configurations, and difficulty keeping track of pipeline runs
- →Collaboration - Barriers collaborating within and across teams
- →Infrastructure - Difficulty managing complex infrastructure and building integrations
Seqera solves these challenges by providing a unified platform for teams to collaborate, configure, monitor, and scale their analyses. This takes us into the modern age of bioinformatics.
3. The Modern Age: Seqera
Compare features of Nextflow and Seqera
The modern age of bioinformatics represents a shift from building everything in-house to leveraging specialized platforms like Seqera that handle infrastructure management. Instead of building infrastructure solutions, teams can focus on their science while Seqera provides the necessary tools for scaling analyses in a unified environment.
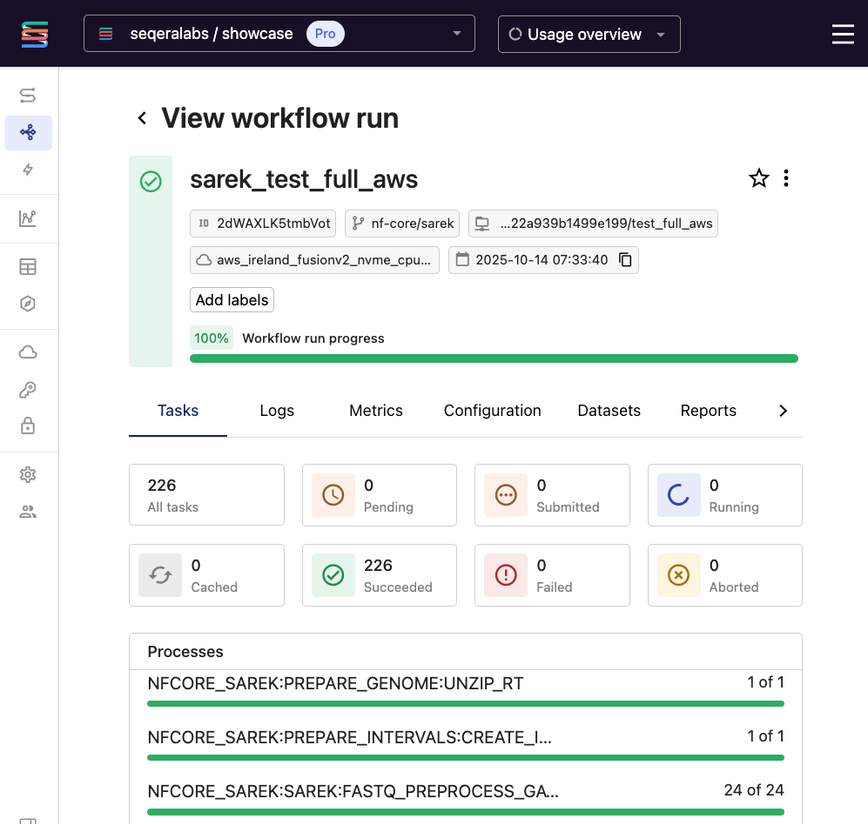
Figure 4. A WGS analysis completed in Seqera with full visibility.
Seqera abstracts infrastructure complexity by unifying data, compute, and containers into collaborative workspaces with organizational guardrails. Additionally, Seqera provides cost optimization through Fusion's container augmentation and supports the entire research journey—from automatically triggering jobs based on data deposits to providing interactive analysis environments through Studios.
One notable aspect is Seqera's seamless journey from development to production. When iterating on Nextflow pipelines, minimal changes are needed to transition new development versions into production environments, supported by comprehensive CI/CD capabilities. We also offer a free tier, complete with compute credits for exploration, making it accessible for teams to evaluate the tooling without initial investment.
Key Advantages
- →Centralized launchpad with a shared environment for teams
- →Real-time monitoring with logs, metrics, and data access
- →Seamless journey from development to production
- →Unified access to your data, code, and compute
However, despite extensive logging, teams often struggle with troubleshooting complex jobs, and navigating multiple configuration layers that are applied to a pipeline run, at both organizational and environment levels. AI tooling addresses these pain points by providing contextual knowledge for rapid iteration. This is why we developed Seqera AI and Seqera MCP.
4. The Agentic Age: Seqera AI and MCP
We’re now entering what could be called the “agentic age” of bioinformatics, characterized by LLMs and MCP servers. Seqera AI (the bioinformatics agent trained in Nextflow, nf-core, and established best practices) leverages LLMs to accelerate workflow iteration and analysis through natural language. With direct integration into Seqera, users can now automatically diagnose stalled pipelines, relaunch workflows with corrected resource limits, and even generate pull requests with new pipeline configurations—all through the AI chat interface.
Try Seqera AI for yourself
Seqera MCP extends these capabilities to allow teams to interact with Seqera via their preferred AI tools (e.g. Claude, Cursor, VS Code Copilot), providing the ability to query both infrastructure and code. This enables improved debugging through contextual analysis of logs and configurations, plus version-controlled pipeline code suggestions that deliver faster solutions to problems. This integration accelerates iterations across code, data, and infrastructure while maintaining the specialized knowledge that makes Seqera uniquely effective for bioinformatics workflows.
Closing Thoughts
Throughout this evolution from the dark ages to today's agentic era, we've seen bioinformatics has consistently advanced by addressing critical bottlenecks and enabling more sophisticated research. Looking toward the future, Seqera AI is positioned to contribute meaningfully to this new era of bioinformatics. What distinguishes Seqera is our foundation built on established tooling: Nextflow, MultiQC, Fusion, and Wave. Having Seqera AI implement solutions based on this robust infrastructure should prove valuable for how effectively teams can iterate on their pipelines.