

Introduction
A wave of advancements, including the generation of massive amounts of data, is transforming how researchers and companies reshape science. Access to compute and workflow management systems is essential to benefit from the new wave of tools. In this blog post, we'll explore how to leverage Seqera Platform and Slurm on Nebius AI Cloud platform using Soperator (Slurm operator for Kubernetes) to deploy scalable Nextflow workflows.
As an example, we will be performing protein structure prediction using the Chai-1 molecular structure prediction method via the nf-chai POC workflow to predict folding of sequences in the Critical Assessment of Techniques for Protein Structure Prediction (CASP15) competition. We’ll dive into:
- →Seqera Platform, an orchestrator for Nextflow, enabling the easy deployment of scalable and reproducible scientific workflows using software containers across all major cloud environments and high performance computing (HPC) environments.
- →Nebius Soperator, a Kubernetes operator that manages Slurm clusters dynamically, bringing Kubernetes' scalability to HPC workloads.
Why Combine Seqera Platform and Nebius Soperator?
Seqera—The best place to run Nextflow
Seqera Platform, developed by the creators of Nextflow, accelerates scientific development by integrating data, code, computing, and environments, empowering researchers to develop, deploy, and scale bioinformatics pipelines faster. Key features include:
- →Centralized pipeline management and monitoring
- →Multi-cloud workflow execution capabilities
- →Built-in versioning and configuration management
- →Reproducible workflow environments
- →Simplified deployment across diverse computing environments
Soperator—The Slurm Operator for Kubernetes
Nebius AI’s Soperator brings modern cloud capabilities to HPC workloads:
- →Automated Slurm cluster provisioning in Kubernetes
- →Easy scaling of Slurm worker nodes to help manage costs
- →Native Docker support for containerized workflows
- →Advanced GPU health monitoring and failure recovery
- →Shared filesystem across cluster nodes
Why this combination?
The integration of Seqera and Soperator creates a powerful environment particularly suited for bioinformatics workflows:
- Seamless Orchestration: Seqera manages workflow logic, while Soperator handles infrastructure
- Full Containerization: Unlike traditional Slurm deployments, Soperator-based Slurm provides native Docker support
- High Availability: Kubernetes' self-healing capabilities ensure workflow reliability
Implementation Guide
Prerequsities:
- →Nebius Cloud account: https://console.eu.nebius.com/
- →Seqera Platform account: seqera.io
- →Basic familiarity with Nextflow and Slurm
Using Soperator to provision a Slurm cluster on Nebius AI
Soperator, Slurm Operator for Kubernetes, bridges the gap between modern cloud-native capabilities of K8s and Slurm's high-performance scheduling, creating a dynamic, scalable HPC solution. Designed to simplify the deployment of Slurm clusters, Soperator offers key features including shared root filesystems, automated health checks, and easy scaling. These ensure automated resource management while maintaining a familiar Slurm user experience.
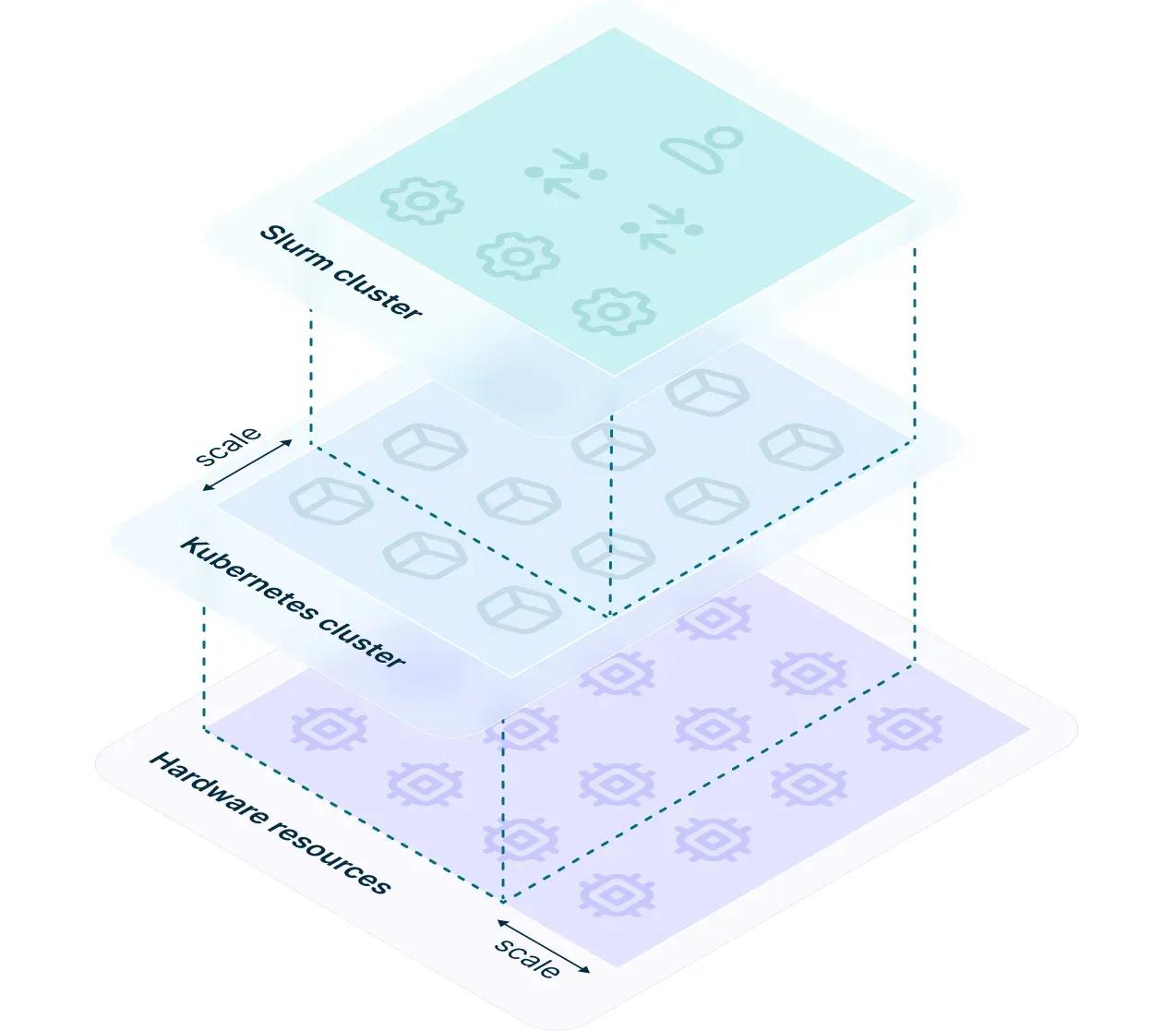
Image source: https://medium.com/nebius/explaining-soperator-nebius-open-source-kubernetes-operator-for-slurm-e7a41f307d14
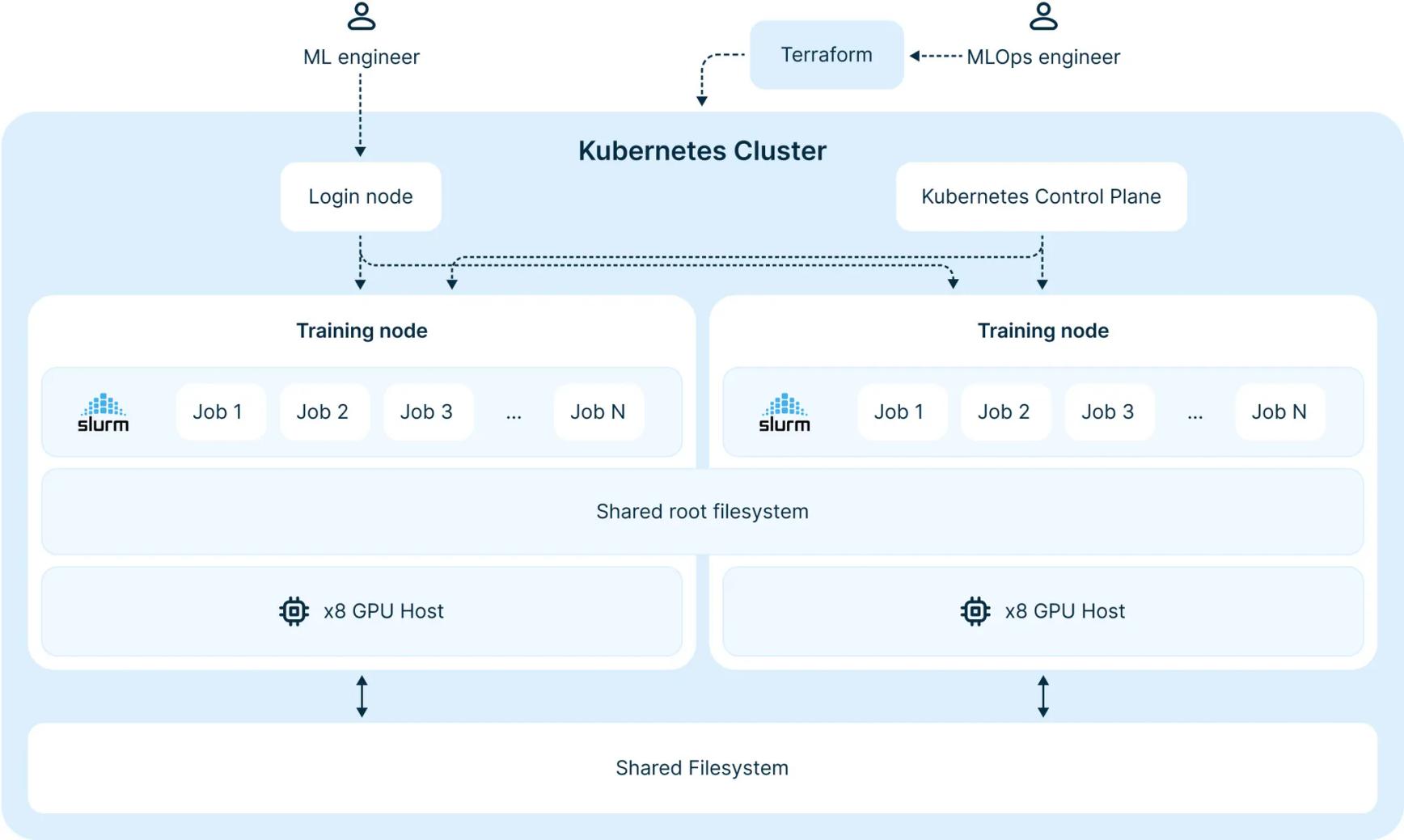
Image source: https://nebius.com/blog/posts/introducing-soperator
Installation Overview
While setting up Soperator is fairly straightfoward, it does require some familiarity with Infrastructure as code (IaC) tools, such Terraform. Below is the summary of the steps you’d need to perform in order to install Soperator on Nebius AI Cloud platform. The detailed step-by-step guide can be found in this GitHub repository.
- Install Terraform, kubectl, and Nebius CLI.
- Define cluster parameters (e.g., compute resources and storage options) in configuration files.
- Use Terraform to configure and deploy your Kubernetes and Slurm clusters.
Alternatively, if you do not wish to follow the above steps, Nebius provides managed Soperator deployments.
Once installed, you can connect with your Slurm cluster via SSH:
Here we can see all GPU nodes available in the Slurm cluster (3 in our test cluster):
Configuring the cluster
- →Creating new Linux users:
This process happens on the shared root filesystem, so you only need to run it once on the login node - the user will be immediately available on all nodes.
- →Installing software packages
Just like with user creation, you only need to run this command once on the login node.
- →Since it is a Slurm cluster, you can submit regular Slurm jobs with
sbatchandsruncommands.
Later on in this blog post we will use Seqera to submit Nextflow pipelines with 1000s of containerized Slurm jobs to the cluster for us.
- →Finally, we will install Java, Nextflow and apptainer on the cluster, since we will require these tools to run Nextflow via Seqera.
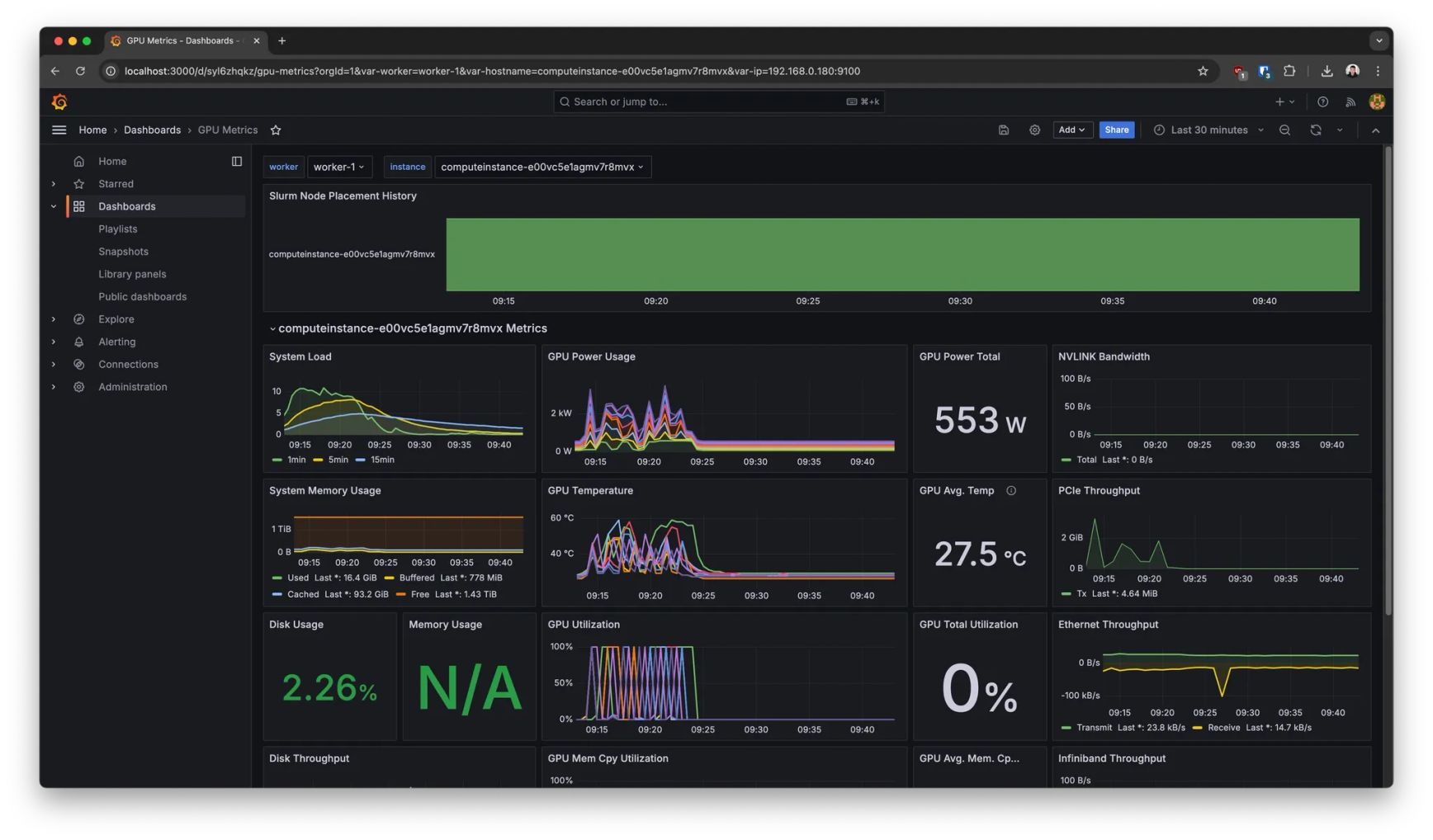
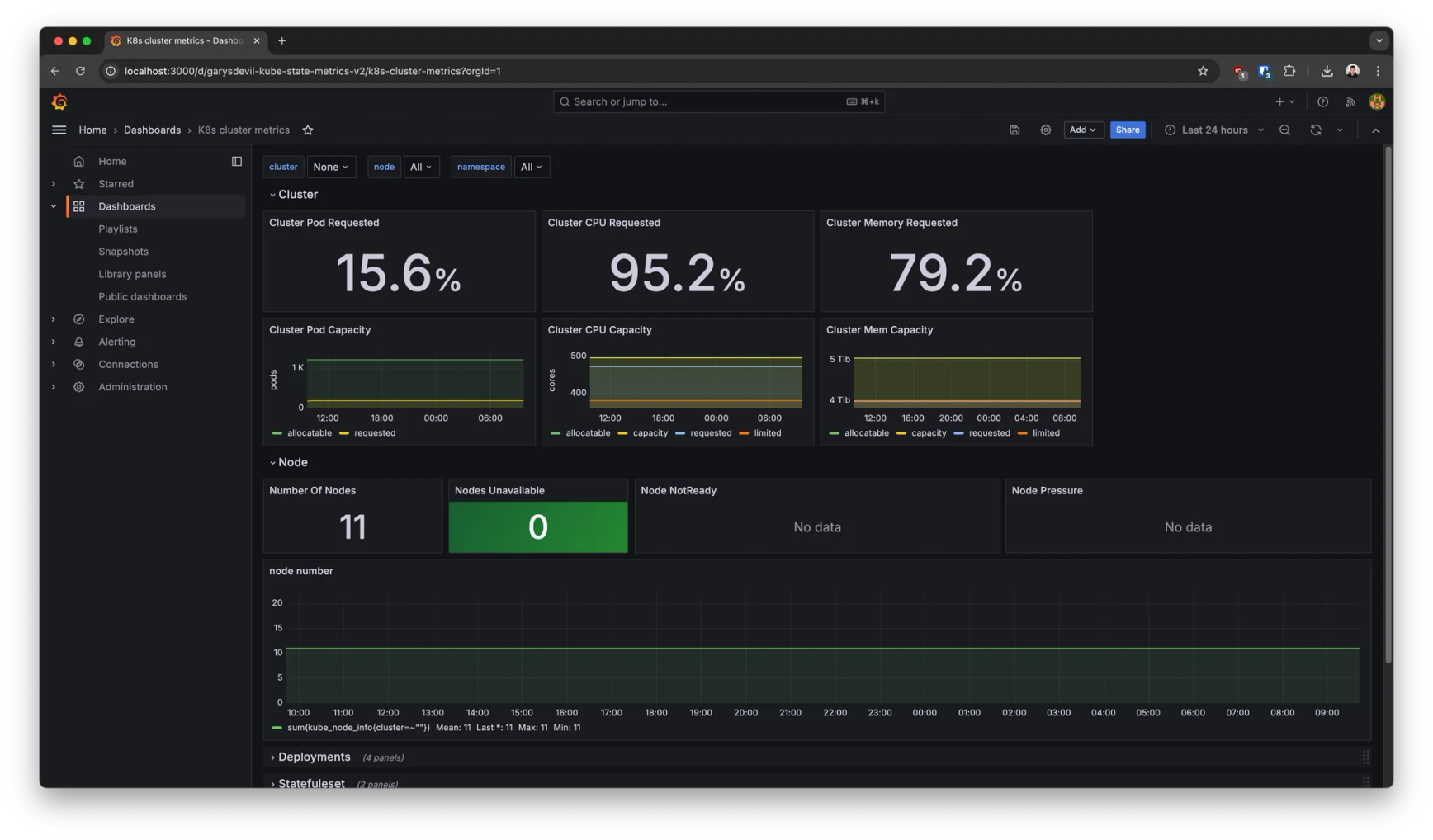
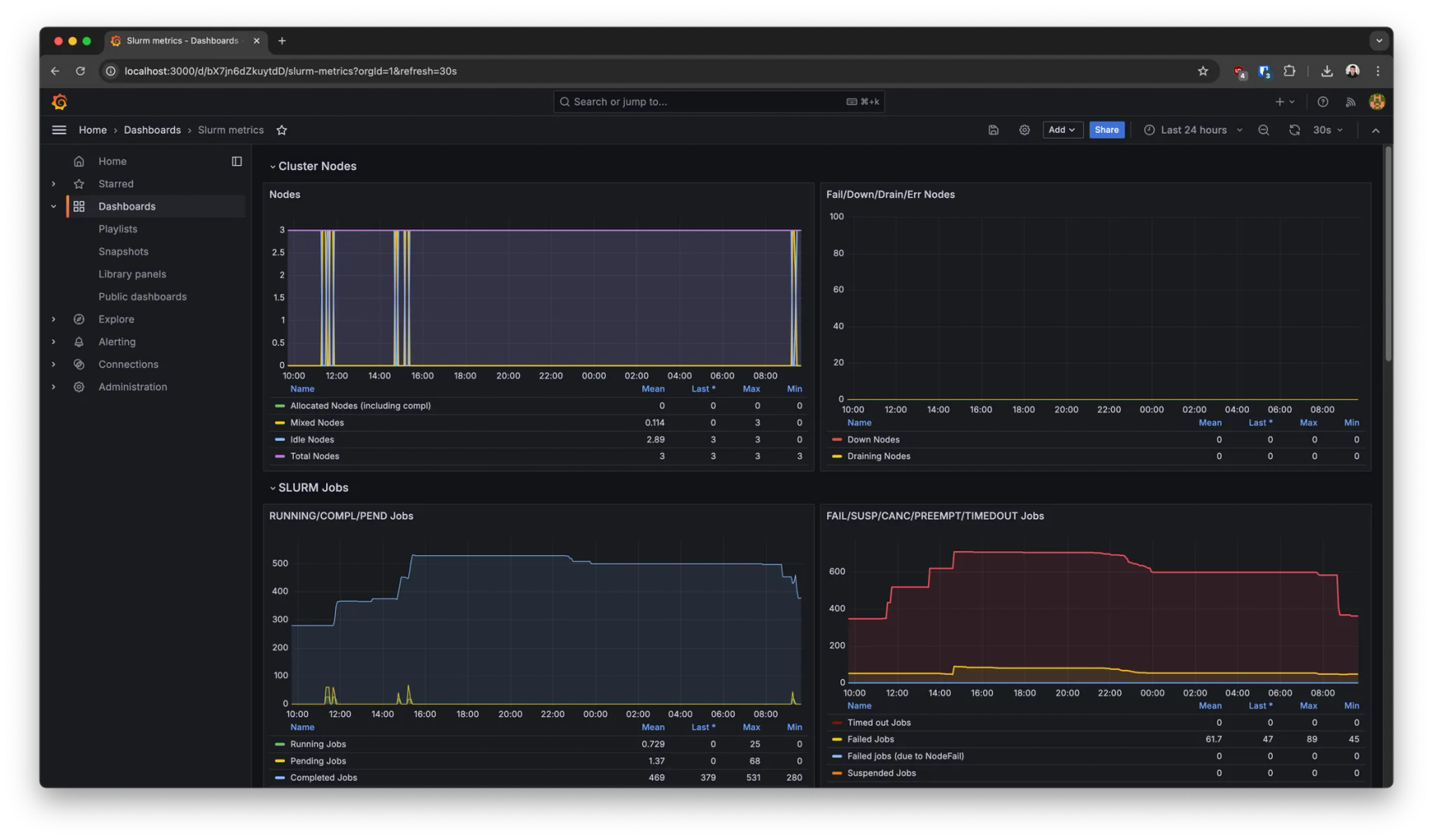
Resource monitoring
This deployment also comes with pre-built dashboards out of the box to monitor, for example:
- →Hardware system metrics (GPU/CPU/RAM/etc utilization)
- →Kubernetes metrics (node availability, pod states. etc)
- →Slurm metrics (worker node states, number of jobs and their status over time, etc).
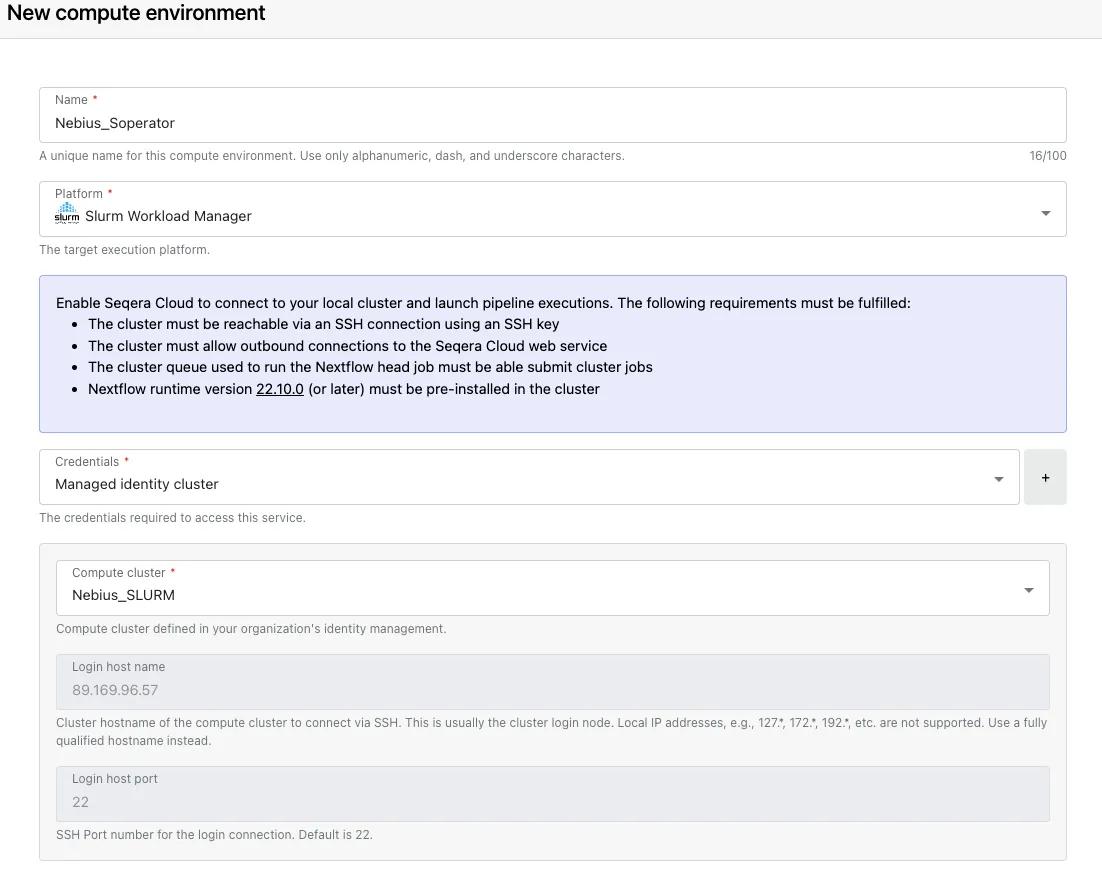
Setting up a Slurm compute environment on Seqera
Now that our Slurm cluster on Nebius is operational and has all necessary tools installed, we can set up access to the Slurm cluster in our Seqera Platform workspace via a compute environment (CE). For detailed information on how to get started with Seqera, please visit this guide. For working with SSH-based Slurm clusters, we recommend setting up managed identities in Seqera to preserve user identity and provide improved security. A detailed walkthrough on how to setup managed identities can be found here: https://docs.seqera.io/platform/24.2/credentials/managed_identities
💡You will need to be the owner of the organization on Seqera to follow the setup guide for managed identities.
Once managed identities are configured, we can create a Slurm CE in our workspace by going to “Compute Environments” —> “Add compute environment” and selecting “Slurm Workload Manager” as the platform and “Managed identity cluster” for credentials. The information for the managed identities will be prefilled for us based on the IP and port information entered under managed identities at the organization level. We specify the work directory where Nextflow will write all temporary work files and specify the head and compute queue names. The head queue will be where our main Nextflow job will run and submit compute jobs to the Slurm scheduler for us. We finish setting up the CE by clicking "Add", which will create the compute environment and bring us back to the main CE page.
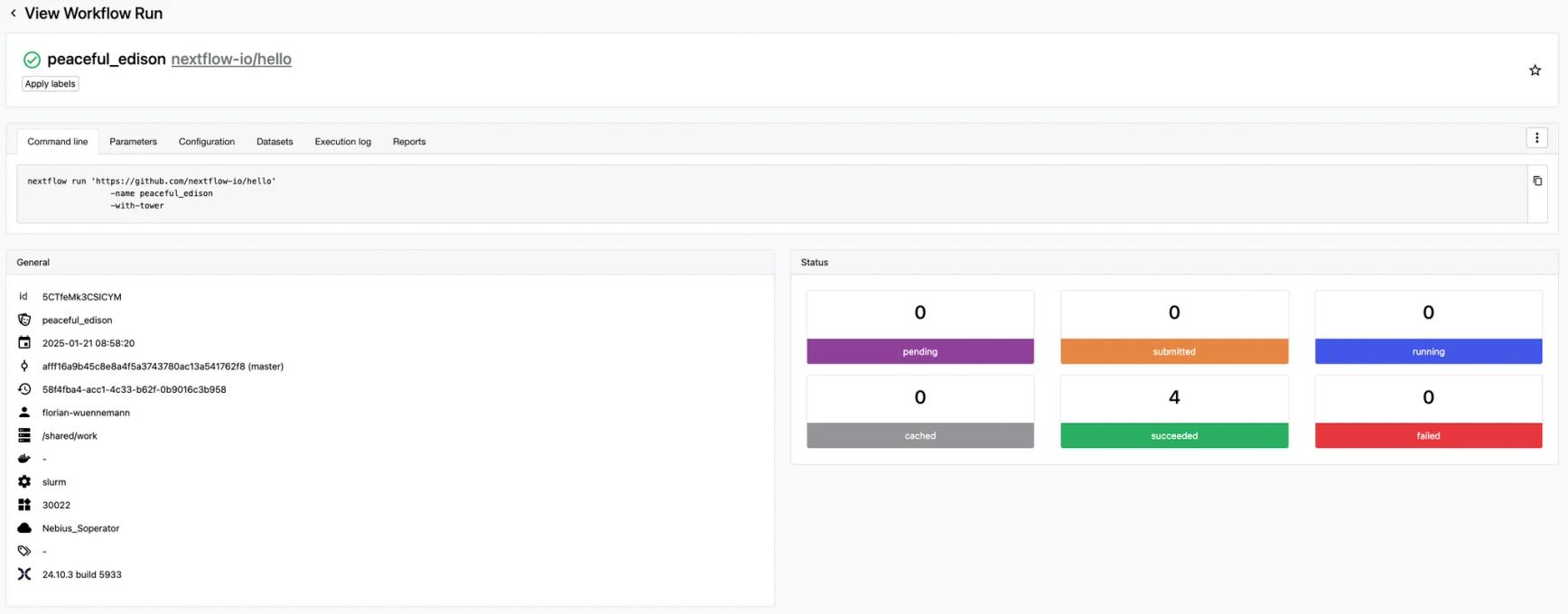
Testing the compute environment
We are now ready to launch Nextflow pipelines on our Soperator Slurm cluster via Seqera. Note that pipeline submission on Seqera can be made via the GUI interface, Seqera Platform API, CLI, or our Python wrapper around the CLI called seqerakit. In this blog post, we will perform all actions via the graphical user interface on seqera.io for simplicity. As a first test, we will run a small test using the nextflow-io/hello pipeline to make sure our setup is working.
In our Launchpad, we click on “Add pipeline”, select our Slurm CE as the compute environment, and add https://github.com/nextflow-io/hello as the pipeline to launch and hit the “Launch” button. This will launch a run of the hello Nextflow pipeline on our Slurm CE. If we go to the run page for this submission, we should see the run kick-off and finish very quickly with 4 succeeding tasks. Great, we are ready to launch real workloads with Nextflow pipelines on our Slurm cluster now.
Running nf-chai on Slurm via Seqera Platform
To showcase compute usage with Seqera and Nextflow on Slurm, we will run a POC pipeline, nf-chai, which implements the state of the art Chai-1 model for molecular structure prediction. Note that this represents a very simple, one process pipeline, which parallelizes running Chai-1 structure prediction in a containerized environment, without any other processes in the pipeline. Nextflow enables you to build data analysis pipelines that can enable data flow through much more complex workflows while providing high scalability.
An example of a more complex pipeline for running multiple structure prediction algorithms is the nf-core/proteinfold pipeline, which is showcased on Seqera here: Protein structure prediction with Seqera. To run Nextflow pipelines on our Slurm cluster, we will use Apptainer as our container engine, as this allows us to easily share apptainer images between nodes.
To efficiently run nf-chai on our Slurm cluster, we will perform two more steps directly on the Slurm cluster. First, we will pre-download the apptainer image to a specified location using the following command:
Next, we will pre-download the reference weights used by Chai-1, to make them available to all workflow tasks. If we don’t do this beforehand, each task would waste time downloading files to the task work directory, instead of symlinking them in, ultimately slowing down our workflow execution. The weights for Chai-1 can be pre-downloaded using the following python command created by a community member on the Chai-1 Github issue board. Make sure to set export CHAI_DOWNLOADS_DIR=[chai1_weights_target_dir] to the directory where you want to store the weights and that you have the chai_lab package installed in your python environment.
Now we are ready to configure and launch nf-chai. Let’s head back to Seqera and add nf-chai as a pipeline from: https://github.com/seqeralabs/nf-chai. For the purposes of this blog post, we will be running release 0.2.1. We need to configure a couple of parameters for a successful execution:
1. Define where we want to store our results (outdir)
2. Point to our pre-downloaded weights dir on the cluster (weights_dir)
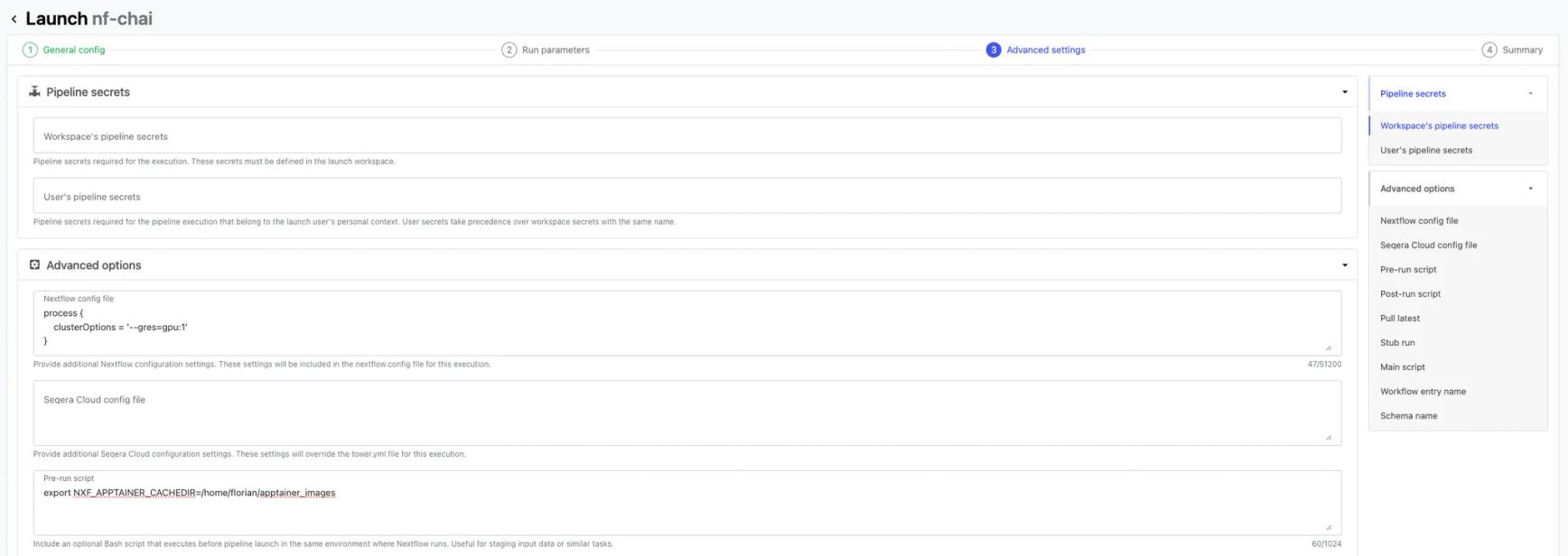
3. Add cluster configuration option to use 1 GPU per task to the Nextflow config block (Advanced settings):
4. Add the apptainer cache dir environment variable to the pre-run script block (“Advanced settings”). This is the location where we downloaded the apptainer image previously.
For input data, we use the CASP15 competition dataset here as an example set of protein sequences to predict using nf-chai. We downloaded the data from CASP15 to our cluster with:
We prepared sequences for prediction by Chai-1 by adding entity type protein| to the fasta IDs and filtered out sequences longer than 2047 amino acids, as Chai-1 cannot currently handle predictions above this size. We run nf-chai using our Soperator Slurm cluster with 3 worker nodes and 8 H100 GPUs per node. Please note that we are running Chai-1 in single sequence mode without multiple-sequence alignment (MSAs) for demonstration purposes here, and prediction quality will likely be lower compared to using Chai-1 with MSAs. We define our input and output data locations for the pipeline run and can finally launch nf-chai. We can now monitor the pipeline starting and jobs going from submitted to running to succeeded status. If we want to, we can also check job status on the Slurm cluster directly with squeue but, we generally don’t need to do this because Seqera will record and report run statistics for us.
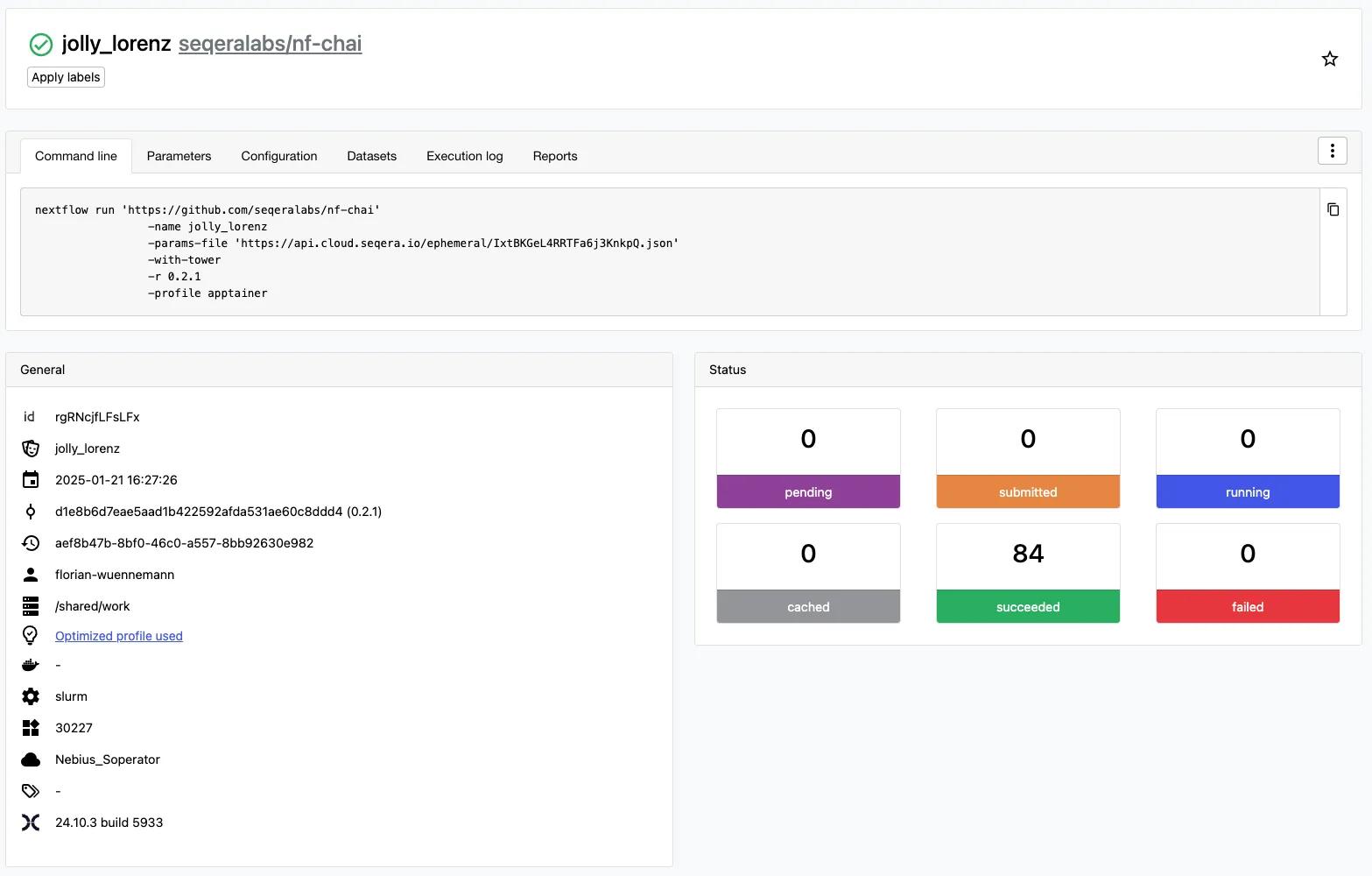
Our nf-chai pipeline run successfully predicted the structures of 84 protein sequences using 24 GPUs in parallel at any given point. We have made all of the results (Chai-1 produces 5 predictions per sequence = 420 structure predictions) available in this Github repository for you to download and explore: https://github.com/seqeralabs/nf-chai-casp15-predictions-blog
3D rendering of protein structure prediction results for target T1105 of CASP15 predicted with Chai-1 via nf-chai.
Conclusion
Combining Nebius’ Soperator with Seqera Platform for deploying your Nextflow pipelines provides great benefits. Soperator-based Slurm provides you with simplified scaling and cluster management experience. Additionally, it is packed with all the necessary drivers and components, making it GPU-ready out of the box. Slurm executor provides a familiar and friendly interface for executing Nextflow workflows and allows easy configuration of compute environments with Seqera. Seqera enables visibility for data processing operations within organizations and facilitates configuration, deployment and optimization of pipelines for execution on Soperator, ensuring stability and reproducibility of your Nextflow runs.
Haven't got a Seqera account? Sign-up now