
This is a joint article contributed to the Seqera blog by Jon Manning of Seqera and Felix Krueger of Altos Labs describing the new nf-core/riboseq pipeline.
In April 2024, the bioinformatics community welcomed a significant addition to the nf-core suite: the nf-core/riboseq pipeline. This new tool, born from a collaboration between Altos Labs and Seqera, underscores the potential of strategic partnerships to advance scientific research. In this article, we provide some background on the project, offer details on the pipeline, and explain how readers can get started with Ribo-seq analysis.
A Fruitful Collaboration
Altos Labs is known for its ambitious efforts in harnessing cellular rejuvenation to reverse disease, injury, and disabilities that can occur throughout life. Their scientific strategy heavily relies on understanding cellular mechanisms via advanced technologies. Ribo-seq provides insights into the real-time translation of proteins, a core process often dysregulated during aging and disease. Altos Labs needed a way to ensure reliable, reproducible Ribo-seq analysis that its research teams could use. While a Ribo-seq pipeline had been started in nf-core, limited progress had been made. Seqera seemed the ideal partner to help build one!
Seqera, known for creating and developing the Nextflow DSL and being an active partner in establishing community standards on nf-core, brought the expertise needed to translate Altos Labs' vision into a viable community pipeline. As part of this collaboration, we formed a working group and also reached out to colleagues at ZS and other community members who had done prior work with Ribosome profiling in Nextflow. Our goal was not only to enhance Ribo-seq analysis capabilities but also to ensure the pipeline’s sustainability through a community-driven process.
Development Insights
The nf-core/riboseq project was structured into several phases:
- →Initial planning: This phase involved detailed discussions between the Scientific Development team at Seqera, Altos Labs, and expert partners to ensure alignment with best practices and effective tool selection.
- →Adapting existing components: Key pre-processing and alignment functions were adapted from the nf-core/rnaseq pipeline, allowing for shareability, efficiency, and scalability.
- →New tool integration: Specific tools for Ribo-seq analysis, such as Ribo-TISH, Ribotricer, and anota2seq, were wrapped into modules using Biocontainers, within comprehensive testing frameworks to prevent regression and ensure reliability. These components were contributed to the nf-core/modules repository, which will now be available for the wider community to reuse, independent of this effort.
- →Pipeline development: Individual components were stitched together coherently to create the nf-core/riboseq pipeline, with its own testing framework and user documentation.
Technical and Community Challenges
Generalizing existing functionality
nf-core has become an encyclopedia of components, including modules and subworkflows that developers can leverage to build Nextflow pipelines. RNA-seq data analysis, in particular, is well served by the nf-core/rnaseq pipeline, one of the longest-standing and most popular members of the nf-core community. Some of the components used in nf-core/rnaseq were not written with re-use in mind, so the first task in this project was to abstract the commodity components for processes such as preprocessing and quantification so that they could be effectively shared by the nf-core/riboseq pipeline.
Test dataset generation
Another significant hurdle was generating robust test data capable of supporting the ongoing quality assurance of our software. In Ribo-seq analysis, the basic operation of some tools depends on the quality of input data, so random down-sampling of variable quality input reads, especially at shallow depths may not be useful to generate test data. To overcome this, we implemented a targeted down-sampling strategy, selectively using input reads that meet high-quality standards and are known to align well with a specific chromosome. This method enabled us to produce a concise yet effective test data set, ensuring that our Ribo-seq tools operate reliably under realistic conditions.
Tool selection
A primary challenge in developing the pipeline was the selection of high-quality, sustainable software. In bioinformatics, funding often limits software development, and many tools are poorly maintained. Furthermore, the understanding of what software 'works' can be ambiguous, embedded in the community's shared knowledge rather than documented formally. Our cooperative approach enabled us to make informed decisions and contribute improvements to the underlying software, enhancing utility for users beyond the nf-core community.
Parameter selection
Selecting the correct parameter settings for optimal operation of bioinformatics tools is a perennial problem in the community. In particular, the settings for the STAR alignment algorithm have very different constraints in Ribo-seq analysis relative to generic RNA-seq analysis. We conducted a series of benchmarks to assess the impact on alignment statistics of various combinations of parameters. We settled on a starting set, but this is a subject of continuing discussion with community members to drive further optimizations.
Pipeline Features
The nf-core/riboseq pipeline is now a robust framework written using the nf-core pipeline template, and specifically tailored to handle the complexities of Ribo-seq data analysis.
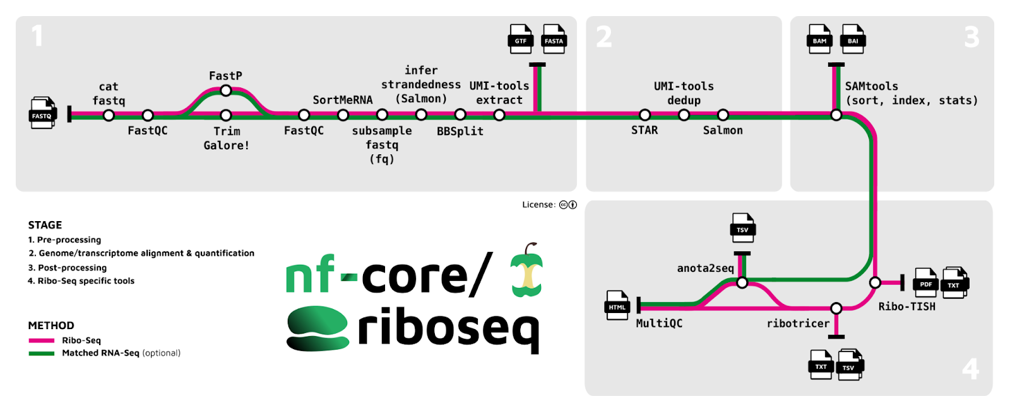
Here is what it offers:
- →Baseline read preprocessing using processes adapted from existing nf-core components.
- →Alignment to references with STAR, producing both transcriptome and genome alignments.
- →Analysis of read distribution around protein-coding regions to assess frame bias and P-site offsets. This produces a rich selection of diagnostic plots to assess Ribo-seq data quality.
- →Prediction and identification of translated open reading frames using tools like Ribo-TISH and Ribotricer.
Assessment of translational efficiency, which requires matched RNA-seq and Ribo-seq data, facilitated by the anota2seq Bioconductor package (see dot plot below).
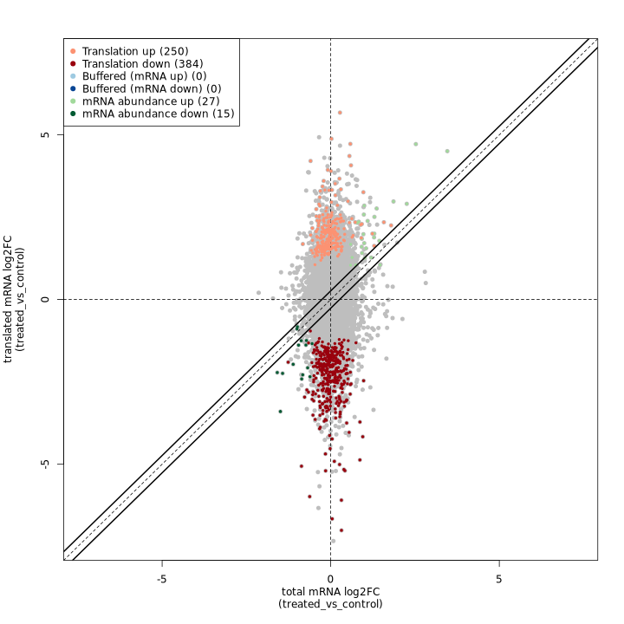
An example result from anota2seq, a tool used to study gene expression, shows how transcription and translation are connected. The x-axis shows changes in overall mRNA levels (transcription) between a treated and a control group, while the y-axis displays changes in the rate of protein synthesis (translation) between those groups, as measured by Ribo-seq. Grey points represent genes with no significant change in either metric and most points align near the center of the x-axis, indicating little change in mRNA levels. However, some genes exhibit increased (orange) or decreased (red) protein synthesis, suggesting direct regulation of translation rather than changes driven solely by mRNA abundance.
If you are a researcher interested in Ribo-seq data analysis, you can test the pipeline by following the instructions in the getting started section of the pipeline. Please feel free to submit bugs and feature requests to drive ongoing improvements. You can also become part of the conversation by joining the #riboseq channel in the nf-core community Slack workspace. We would love to see you there!
Next Steps
Following this initial phase of work, Seqera and Altos Labs have handed over the nf-core/riboseq pipeline to the nf-core community for ongoing maintenance and development. As members of that community, we will continue to play a part in enhancing the pipeline going forward. We hope others will benefit from this effort and continue to improve and refine pipeline functionality.
Coincidentally the authors of riboseq-flow published their related work on the same day that nf-core/riboseq was first released. This pipeline has a highly complementary set of steps, and there is already ongoing collaboration to work together to build an even better community resource.
Empowering Research and Innovation
The joint contribution of Seqera and Altos Labs to the nf-core/riboseq pipeline highlights how collaboration between industry and open-source communities can result in tools that push scientific boundaries and foster community engagement and development. By adhering to rigorous code quality and testing standards, nf-core/riboseq ensures researchers access to a dependable, cutting-edge tool.
We believe this new pipeline is poised to be vital in studying protein synthesis and its implications for aging and health. This is not just a technical achievement - it's a step forward in collaborative, open scientific progress.
If you have a project in mind where Seqera may be able to help with our Professional Services offerings, please contact us at services@seqera.io. We are the content experts for Nextflow, nf-core, and the Seqera Platform, and can offer tailored solutions and expert guidance to help you fulfill your objectives.
To learn more about Altos Labs, visit https://www.altoslabs.com/.
Acknowledgments
nf-core/riboseq was initially written by Jonathan Manning (Bioinformatics Engineer at Seqera) in collaboration with Felix Krueger and Christel Krueger (Altos Labs). The development work carried out on the pipeline was funded by Altos Labs. We thank the following people for their input (in alphabetical order):
- →Felipe Almeida (ZS)
- →Anne Bresciani (ZS)
- →Caroline Eastwood (University of Edinburgh)
- →Maxime U Garcia (Seqera)
- →Mikhail Osipovitch (ZS)
- →Jack Tierney (University College Cork)
- →Edward Wallace (University of Edinburgh)