
Learn how to run your first analysis pipeline
Run your first analysis pipeline in under 30 minutes
Nextflow continues to grow in popularity. Today, the open-source workflow manager is used by thousands of bioinformaticians, clinicians, and data scientists and is downloaded 160,000+ times monthly. Despite this, many Nextflow users are unfamiliar with Tower, a graphical interface for the Nextflow platform.
Based on data from our April 2023 State of the Workflow Survey, over 50% of Nextflow users are unfamiliar with Tower. This is despite the fact that 75% view the ability to monitor pipeline runs using a graphical interface as important or very important. 1
If you fall into this category, you’re in luck! This guide provides a short Tower tutorial and walks you through the process of running your first pipeline. The steps in this guide can be completed in under 30 minutes.
Watch the Getting Started with Tower Video
About Tower Cloud
Tower is an intuitive interface that enables users of all skill levels to quickly launch, monitor, and manage scalable Nextflow pipelines. While the interface is excellent for enabling non-technical users, it can also provide a significant productivity boost for experienced bioinformaticians and clinicians. With Tower, users can:
- →Collaborate and share data securely among local and remote teams.Easily share pipelines, prior runs, and compute environments.
- →Run pipelines in any environment, from workstations to private, hybrid, and public clouds.
- →Reduce cloud spending with powerful resource optimization features.
- →Simplify troubleshooting by consolidating information about pipeline runs in a single interface.
There are multiple Tower offerings, ranging from a free version of Tower Cloud to Tower Cloud Professional to self-hosted Tower Enterprise.
Getting started
1. Obtain a free Tower Cloud account
Tower Cloud does not require a new password or registration procedure. Users can simply navigate to https://cloud.tower.nf, and click Sign In. You can access Tower using existing GitHub or Google credentials or by providing an e-mail address.
2. Familiarize yourself with the Community Showcase
After logging into Tower Cloud, you will be directed to the Community Showcase workspace. Workspaces in Tower enable sharing pipelines, runs, and environments with other users. The Community Showcase contains predefined pipelines, datasets, and compute environments that you can use to familiarize yourself with the Tower environment.
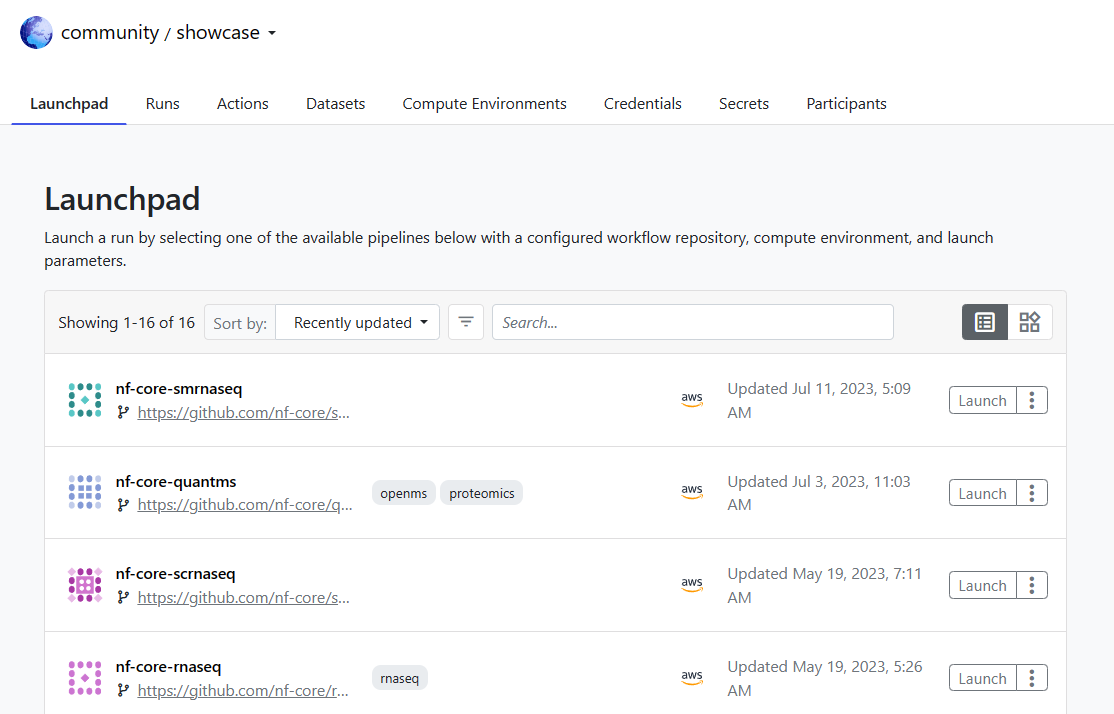
The Launchpad tab is pre-populated with several popular nf-core pipelines. nf-core is a community effort to curate and share high-quality analysis pipelines.
Select the Datasets tab to familiarize yourself with the data pre-loaded into the showcase. Datasets are essentially metadata, formatted as versioned TSV or CSV format files. The physical files associated with Datasets typically reside in a cloud object store such as Amazon S3.
3. Review the Compute Environments
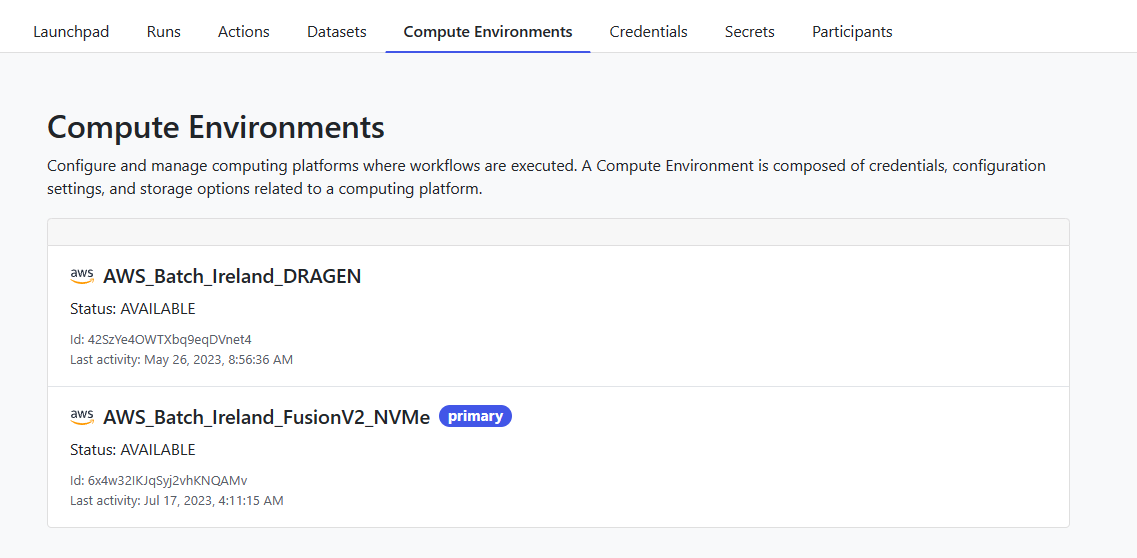
Before launching a pipeline, it is instructive to navigate to the Compute Environments tab. A Compute Environment comprises credentials, configuration settings, and storage options related to a specific computing platform. Compute Environments can be accessed by all users in a shared workspace.
Typically, users provide their own cloud credentials or details about on-premises HPC clusters in a Compute Environment. Tower supports most popular cloud-based compute environments, including AWS Batch, Azure Batch, Google Cloud Batch, and Kubernetes. Default Compute Environments are pre-configured in the Community Showcase, enabling users to experiment with Tower for free.
Read the article Nextflow on BIG IRON for additional information on supported HPC clusters
You can click on the name of a Compute Environment to see how it is configured.
4. Launch a pipeline
To launch your first pipeline, navigate to the Launchpad tab in the Community Showcase. The nf-core-rnaseq pipeline is an excellent place to start. nf-core/rnaseq analyzes RNA sequencing data obtained from organisms with a reference genome and annotation. It is one of the most popular nf-core analysis pipelines, used by thousands of bioinformaticians worldwide.

Tower will guide you through the process of entering pipeline parameters. Default values and instructions are provided for each field. Depending on the pipeline, Tower exposes rich configuration options that enable you to control the behavior of pipelines at runtime. To get started, you can accept the default parameters.
Most production-quality Nextflow pipelines are Tower ready. A nextflow_schema.json file in the pipeline’s code repository facilitates the intuitive guided interface.
See Best Practices for Deploying Pipelines with Nextflow Tower
The sample community pipelines are stored in GitHub, but Tower supports most Git-compatible code repositories, including GitLab and BitBucket.
Click the Launch button at the bottom of the screen to launch the pipeline with your selected parameters.
5. Monitor pipeline execution
After launching a pipeline, you will be redirected to the Runs tab. The showcase is a shared environment so you will see your runs as well as pipelines launched by other users. Tower will automatically assign a name to your pipeline and any Labels you might have specified when submitting the pipeline. You can search the pipeline run history by entering a value such as label:rnaseq in the search field.
Click on the name of the submitted pipeline to monitor its execution. Tower exposes details about each pipeline, as shown:
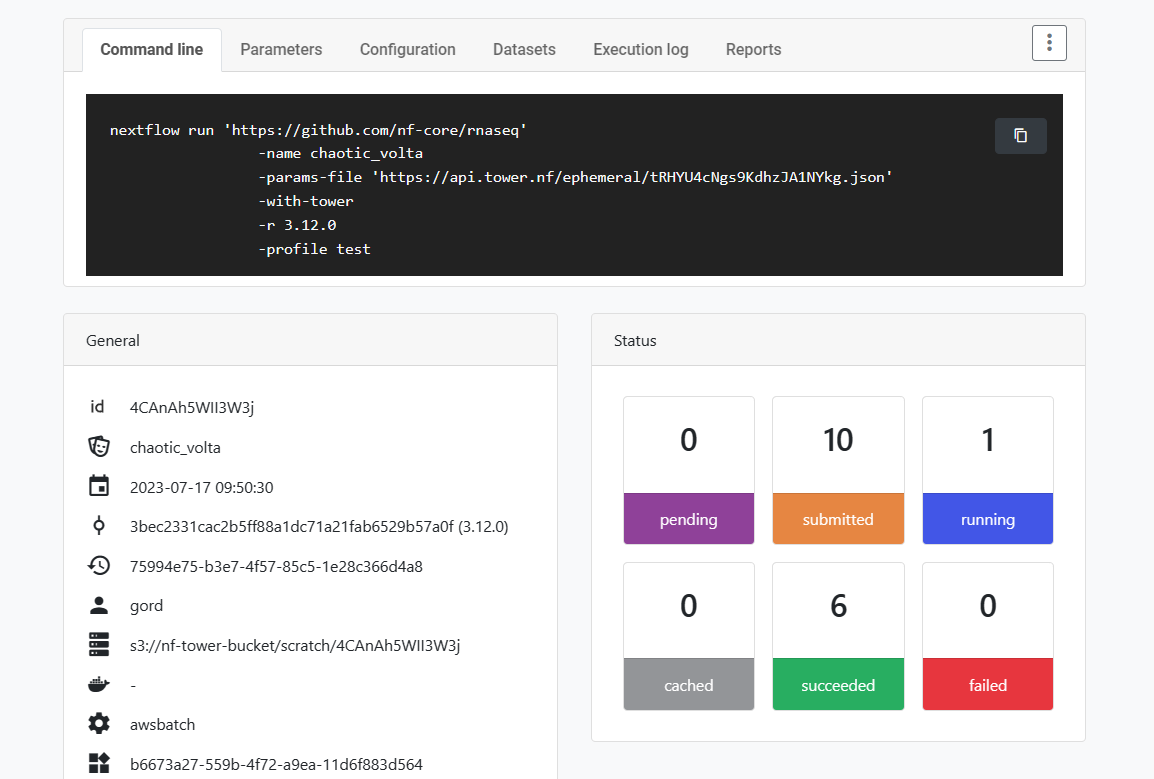
The default compute environment will automatically provision virtual machines on AWS Batch when a pipeline is launched. As a result, it will take a minute or two for execution to begin.
As the pipeline runs, you can scroll through the Tower interface and monitor execution. Tower exposes all details, including data files, log files, and task-level execution details. You can even monitor the pipeline’s Execution log in real-time.
The beauty of running pipelines in Tower is that colleagues in a shared workspace can access all the same detailed information for each run, simplifying collaboration on scientific workflows and datasets.
6. View the results
Once the pipeline completes, additional information will be available under the Runs tab, including interactive charts detailing CPU and Memory usage, Job durations, and I/O activity. By analyzing these metrics, Tower can suggest optimizations that will enable the pipeline to run more efficiently. These optimizations have been shown to reduce resource requirements and associated spending by up to 87%.2
See the article optimizing resources usage using Nextflow Tower
Most Nextflow pipelines will generate reports or output files which are useful to inspect at the end of the pipeline execution. Instructions on how to make reports accessible through Tower are provided in the Tower documentation. For pipelines that support reports, execution results will be available under Reports tab for each workflow run.
The nf-core/rnaseq pipeline organizes output using MultiQC. You can select the MultiQC HTML report under the Reports as shown below, and open MultiQC in a new tab to view results interactively.
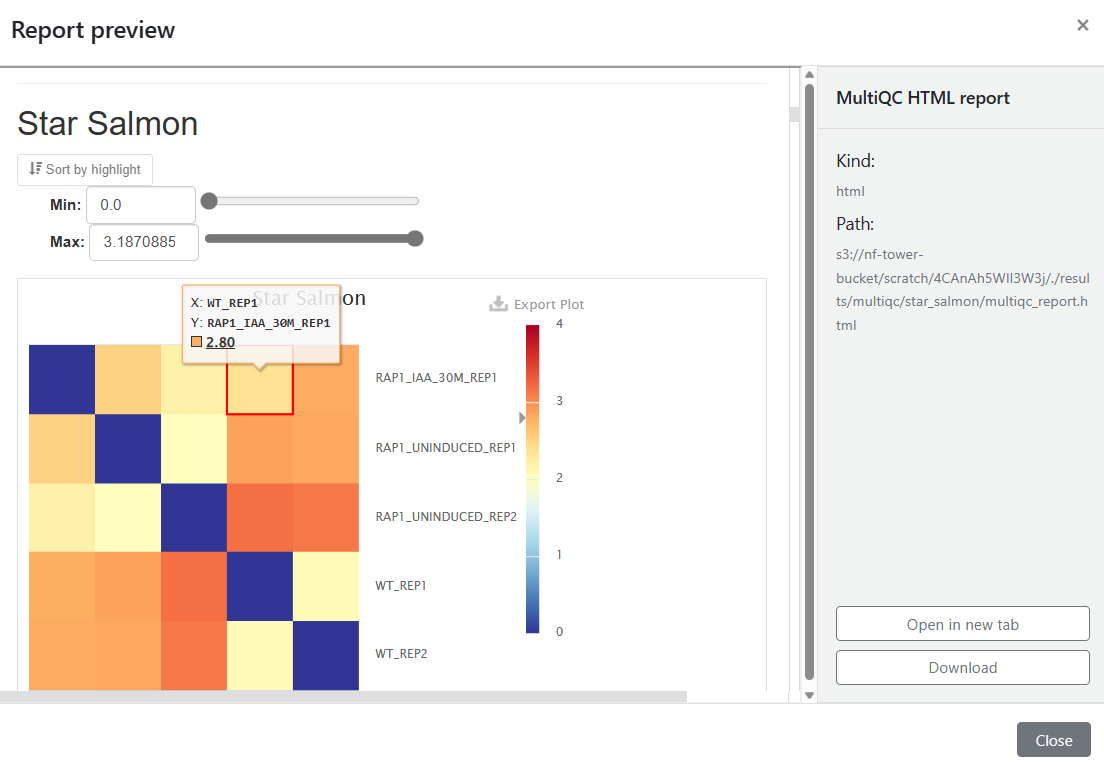
You can learn more about MultiQC at https://multiqc.info.
7. Next steps
Congratulations, if you’ve made it this far, you have successfully run your first pipeline in Tower.
You can navigate to your personal workspace and add your own compute environments and pipelines. You can also create new private organizations and workspaces and invite colleagues to join you in Tower Cloud.
Consult the Tower Documentation to learn more.
Watch the YouTube video for an accelerated Introduction to Tower Cloud.
1 In the survey, when asked about the importance of the ability to monitor pipeline runs via a user interface (Question 32) 35.91% viewed this capability as very important, 36.53% viewed it as important, and 18.16% were unsure. The remaining 9.4% viewed this capability as unimportant. Based on a survey of 502 Nextflow users.
2 The nf-core/viralrecon pipeline was used for this test. The estimated pipeline cost before optimization was $9.97, and the cost was reduced to $1.54 with Tower optimization. See details in Optimizing resource usage with Nextflow Tower.