
As bioinformaticians, reproducibility becomes almost an obsession. We need to ensure that the analysis we perform today, with the same input data, will yield the same results tomorrow. Even more critically, these results should remain consistent across different machines or infrastructures. Over the years, we've made significant strides toward achieving this goal, and at Seqera, we’re at the forefront of these efforts. With a recent feature in Wave, bioinformaticians and data scientists alike are about to experience an important step forward. But before we dive into that, let’s take a step back and explore some context.
The evolution of bioinformatics environments
In the early days, bioinformatics analyses were typically run on single machines, with software either installed system-wide by administrators or individually by users in their personal environments. This led to problems when someone upgraded R or Python, and suddenly, any results reliant on the older version were no longer reproducible.
As the field evolved, virtual environments became essential. They allowed analyses to be carried out in isolated environments with well-defined software requirements, making it possible to recreate results precisely at later dates. Conda made this even simpler, managing dependencies across disparate software ecosystems. More recently, container platforms like Docker have further streamlined reproducibility by packaging entire environments—including software, dependencies, and the operating system—into portable containers. This ensures analyses run consistently across different systems and infrastructures. Docker and similar platforms have become key tools for sharing work that can be reliably reproduced, regardless of the computational setup.
Tools like Nextflow took this a step further, allowing us to define independent software environments for every process in a workflow—sometimes hundreds of steps. This solved many dependency conflicts and made reproducibility more achievable. The nf-core community, for example, has built an expanding library of nearly 1,500 individually reusable workflows and modules. These modules come with predefined software for Conda, Docker and Singularity. However, one challenge we've encountered is ensuring replicability when the same software is deployed via different methods, particularly between Conda and containers. This issue largely stems from differences in what are known as “transitive dependencies.”
Transitive dependencies and the Conda-Docker gap
To illustrate, let’s consider a real-world example of a Conda environment configuration file used by an nf-core module:
This configuration specifies that the process requires version 1.34.0 of the DESeq2 R package. It’s hosted in the Bioconda repository, and thanks to the magic of the BioContainers infrastructure, there’s also a Docker container available at quay.io. This means we can run the same process with identical versions of the DESeq2library, both inside and outside a container runtime.
This setup seems ideal—and, initially, it likely works as expected.However, the Conda package, bioconda::bioconductor-deseq2=1.34.0, has its own set of dependencies within the Bioconda repository and other Conda channels. When running a Nextflow workflow with Conda, the environment is resolved based on the package ecosystem at the moment the workflow is run, potentially pulling newer versions of dependencies, including crucial components like R itself. Over time, this can cause freshly built Conda environments to diverge from the Docker container that was frozen in time, undermining reproducibility and potentially leading to unexpected failures in tests, such as nf-test snapshots. Investigating these changes can require some serious detective work inside the container.
The case for environment locking
At this point, you might wonder: “Why not just record the version of every package used?” This is indeed where we’re heading—what we refer to as “locking” the environment. Rather than replacing flexible environment files, which are valuable for new projects needing the latest dependencies, we aim to provide lockfiles alongside these configurations. These lockfiles will match the versions used in Docker containers, ensuring exact (or nearly-exact) reproducibility of behaviors across different systems.
Enter Wave
This is where Wave comes in. An open-source tool from Seqera, Wave streamlines the process of generating containers for workflows, offering several advantages over the Biocontainers, especially when building containers for multiple Conda packages or when using Singularity.
With the Wave command-line interface (CLI) or the Seqera Containers web interface, you can request a container (Docker or Singularity) for a specified system architecture (arm64, amd64) based on a Conda environment:
Wave builds the environment and provides the container.
Wave and lockfiles: replicable behavior inside and outside containers
Until now, that was the end of the process: Wave built your container. But a year later, a new Conda environment built with the same specification would likely differ from this specific container's environment. A recent innovation in Wave now addresses this: along with building containers, Wave also preserves the Conda environment by running the following command internally:
This generates a lockfile for every container. A lockfile for the DESeq2 example above might look like this:
With this feature, Wave fixes the versions of ALL the Conda packages in the environment. But it doesn’t just pin down dependencies using version numbers like before—it creates an explicit set of links to the Conda package files themselves. This means that the slow process of resolving dependencies when building an environment is also bypassed entirely. Using this lockfile in workflows, you not only get more reproducible results with Conda but faster ones too. Note that the URLs also include md5sum for the package downloads, ensuring an absolutely faithful environment.
Hands-on with Wave and Conda lockfiles
So how do you use Wave to generate a paired container definition and Conda lockfile? You have two choices here:
- Seqera Containers: A web interface that provides easy access to Wave's functionality.
- Wave CLI: For those comfortable with the Linux command line.
Seqera Containers
Seqera Containers is a web resource which gives you easy access to Wave functionality via a graphical interface. In the example below we select the R-based Bioconda package ‘r-shinyngs’ and hit ‘Get Container’.
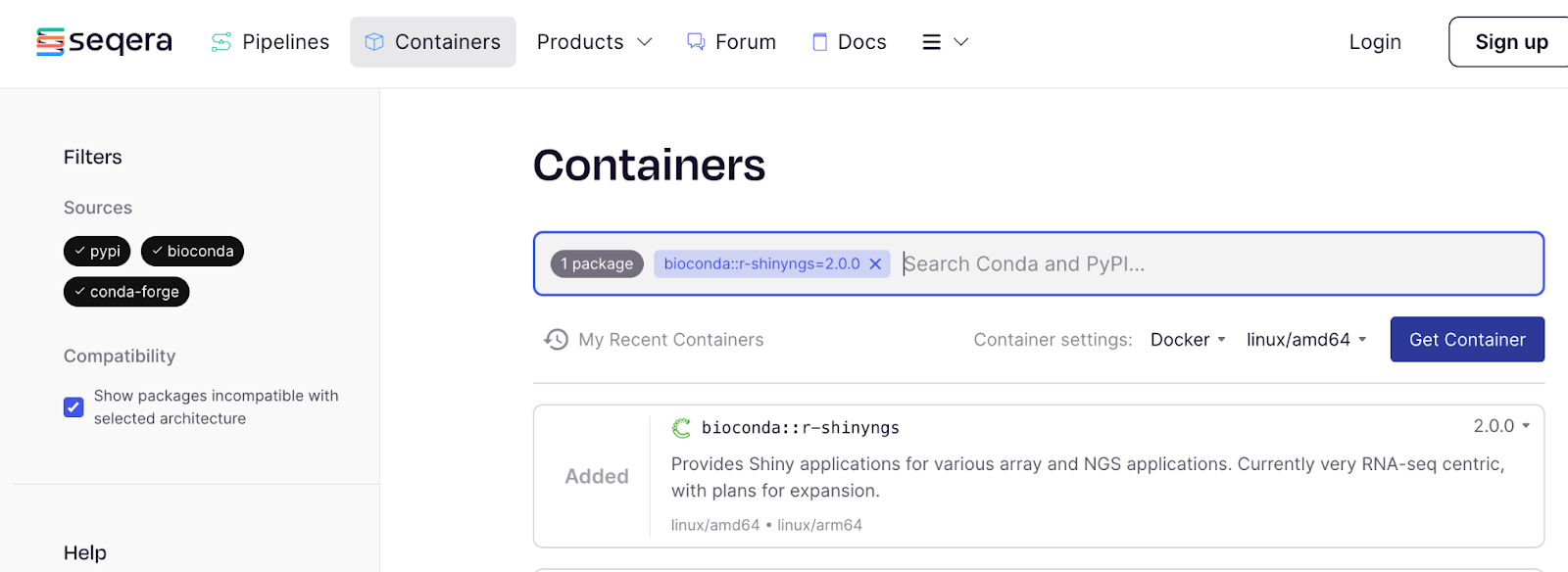
That gives us a pop-up with the link to the container, which we can make a note of:
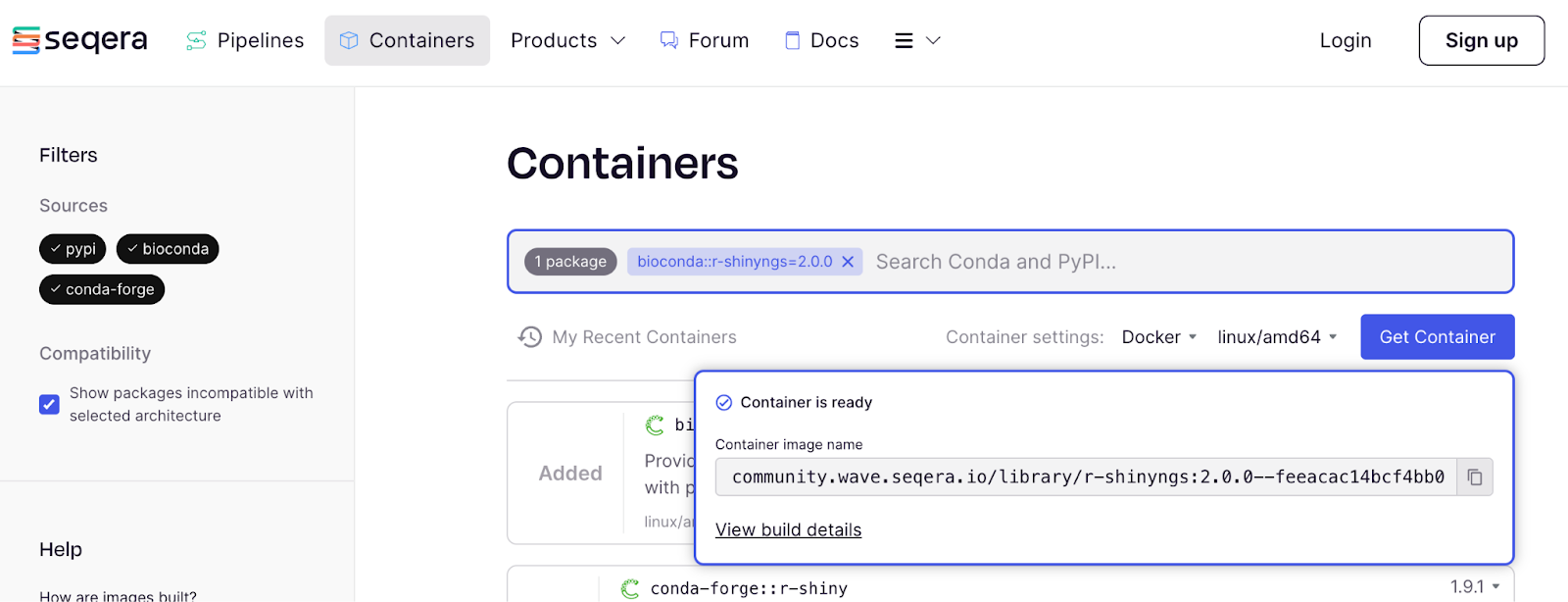
If we then click ‘View build details’:
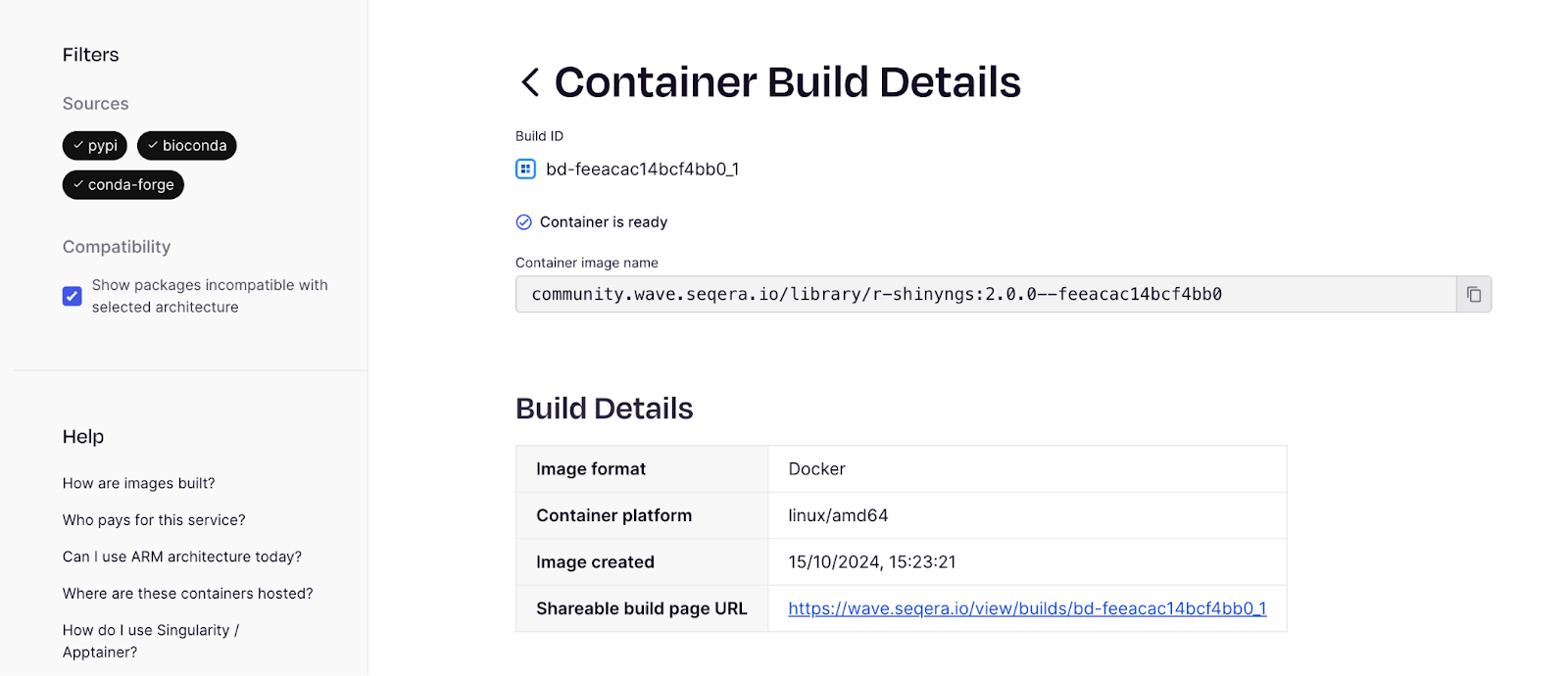
… and then the ‘Sharable build page URL’:
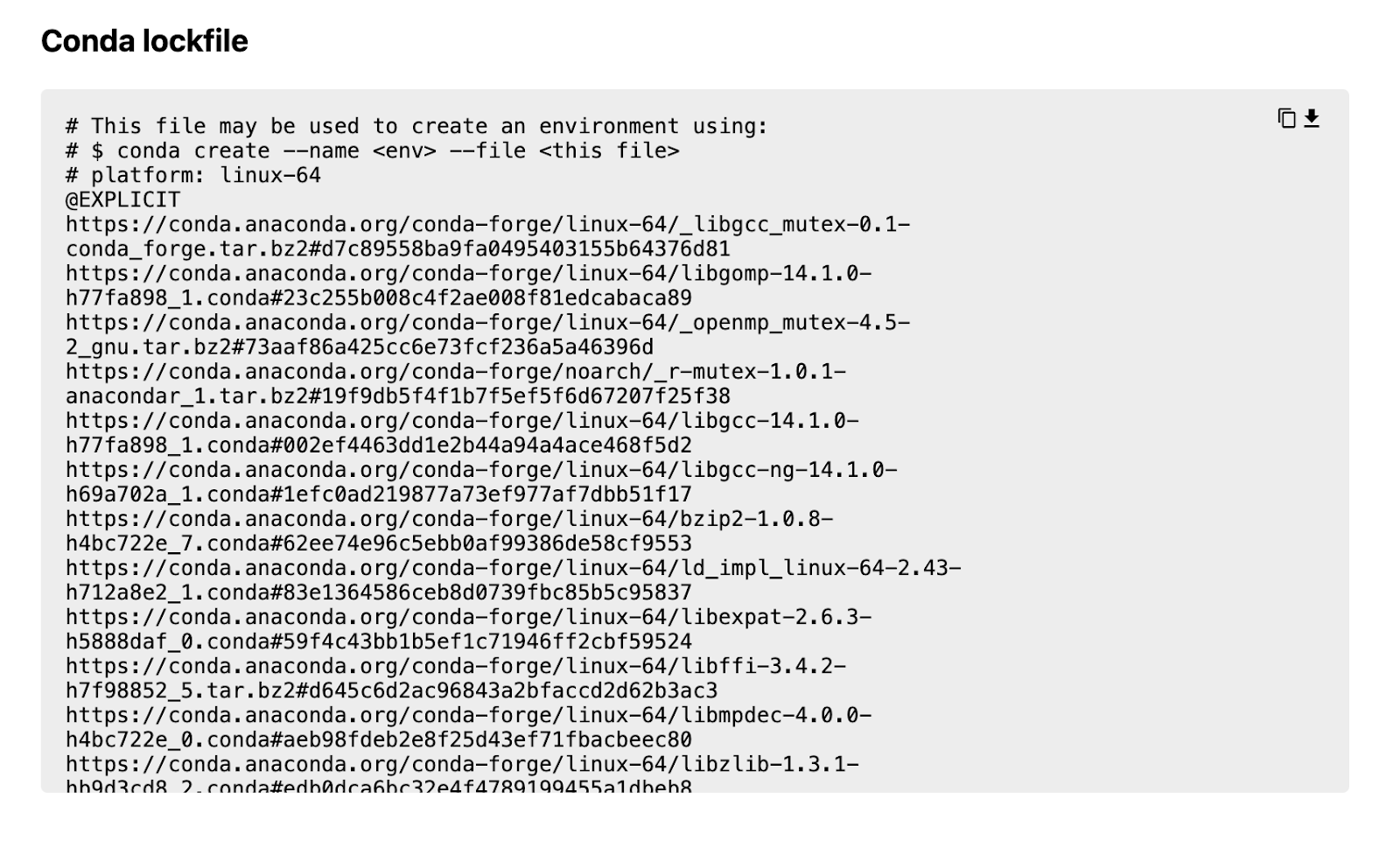
This gives us access to a page with the lockfile available via a text box with a download button.
The Wave CLI
If you’re used to working from the Linux command line, the Wave CLI allows you to get both the container and Conda lockfile for a given package specification in just a couple of commands.
First, run Wave with your Conda package specification (see the documentation linked above for more information):
Be sure to specify the correct platform, and the yaml output option. This will give you a result like:
The ‘targetImage’ field in the output gives you an image you can use straight away. The URI for the Conda lockfile can be generated from the buildId:
Testing the container and Conda lockfiles
To show the comparability of the Docker and Conda approaches, we can use both software definitions to derive the version of a transitive dependency, something whose version we didn’t explicitly fix.
To do using the container we obtained via Wave:
This gives us output like:
To do same in a Conda environment built from the lockfile, we must download the lockfile and use it to build a Conda environment, before running the command:
The environment creation is very fast, because we’re bypassing dependency resolution. The output of the R command is:
This matches what we found in the container.
Transparency and portability
Docker has long been used to “freeze” software environments, but it’s not always transparent—especially when it comes to querying the versions of transitive dependencies. Containers work well as delivery mechanisms, but by preserving Conda environments separately, we get the best of both worlds: the convenience of containers for provisioning software (particularly in cloud environments) and the transparency and reproducibility of Conda lockfiles. These lockfiles provide an explicit record of all dependency versions, ensuring a near-identical environment both inside and outside container runtimes. If we ever lose sight of the container for whatever reason, we can also rebuild something almost identical based on that lockfile.
With Wave, reproducibility becomes faster, easier, and more consistent across platforms. We're excited to continue pushing the boundaries of bioinformatics workflows, helping you achieve the same results, every time, everywhere.
💡Interested in learning more about Wave? Download the whitepaper now