
The ability to resume an analysis (i.e. caching) is one of the core strengths of Nextflow. When developing pipelines, this allows us to avoid re-running unchanged processes by simply appending -resume to the nextflow run command. Sometimes, tasks may be repeated for reasons that are unclear. In these cases it can help to look into the caching mechanism, to understand why a specific process was re-run.
We have previously written about Nextflow's resume functionality as well as some troubleshooting strategies to gain more insights on the caching behavior.
In this post, we will take a more hands-on approach and highlight some strategies which we can use to understand what is causing a particular process (or processes) to re-run, instead of using the cache from previous runs of the pipeline. To demonstrate the process, we will introduce a minor change into one of the process definitions in the the nextflow-io/rnaseq-nf pipeline and investigate how it affects the overall caching behavior when compared to the initial execution of the pipeline.
Local setup for the test
First, we clone the nextflow-io/rnaseq-nf pipeline locally:
In the examples below, we have used Nextflow v22.10.0, Docker v20.10.8 and Java v17 LTS on MacOS.
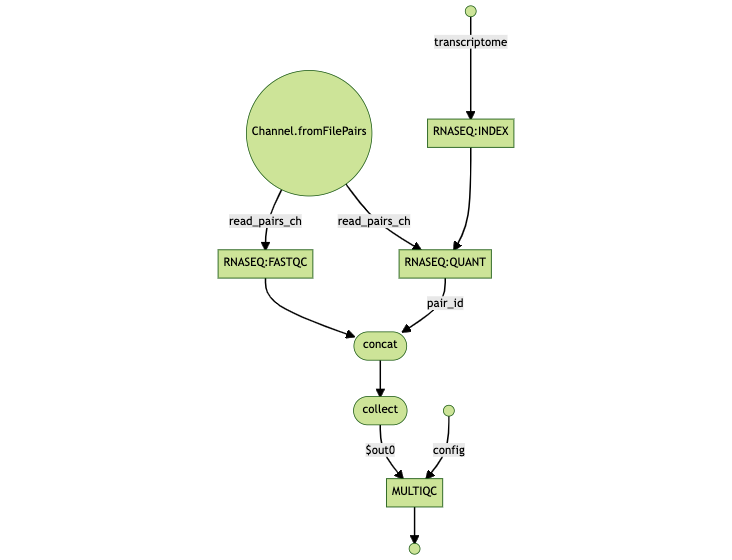
Pipeline flowchart
The flowchart below can help in understanding the design of the pipeline and the dependencies between the various tasks.
Logs from initial (fresh) run
As a reminder, Nextflow generates a unique task hash, e.g. 22/7548fa… for each task in a workflow. The hash takes into account the complete file path, the last modified timestamp, container ID, content of script directive among other factors. If any of these change, the task will be re-executed. Nextflow maintains a list of task hashes for caching and traceability purposes. You can learn more about task hashes in the article Troubleshooting Nextflow resume.
To have something to compare to, we first need to generate the initial hashes for the unchanged processes in the pipeline. We save these in a file called fresh_run.log and use them later on as "ground-truth" for the analysis. In order to save the process hashes we use the -dump-hashes flag, which prints them to the log.
TIP: We rely upon the `-log` option in the nextflow command line interface to be able to supply a custom log file name instead of the default .nextflow.log.
Edit the `FastQC` process
After the initial run of the pipeline, we introduce a change in the fastqc.nf module, hard coding the number of threads which should be used to run the FASTQC process via Nextflow's `cpus` directive.
Here's the output of git diff on the contents of modules/fastqc/main.nf file:
Logs from the follow up run
Next, we run the pipeline again with the -resume option, which instructs Nextflow to rely upon the cached results from the previous run and only run the parts of the pipeline which have changed. As before, we instruct Nextflow to dump the process hashes, this time in a file called resumed_run.log.
Analysis of cache hashes
From the summary of the command line output above, we can see that the RNASEQ:FASTQC (FASTQC on ggal_gut) and MULTIQC processes were re-run while the others were cached. To understand why, we can examine the hashes generated by the processes from the logs of the fresh_run and resumed_run.
For the analysis, we need to keep in mind that:
- →The time-stamps are expected to differ and can be safely ignored to narrow down the `grep` pattern to the Nextflow `TaskProcessor` class.The _order_ of the log entries isn't fixed, due to the nature of the underlying parallel computation dataflow model used by Nextflow. For example, in our example below, `FASTQC` ran first in `fresh_run.log` but wasn’t the first logged process in `resumed_run.log`.
Find the process level hashes
We can use standard Unix tools like grep, cut and sort to address these points and filter out the relevant information:
- →Use `grep` to isolate log entries with `cache hash` stringRemove the prefix time-stamps using `cut -d ‘-’ -f 3`Remove the caching mode related information using `cut -d ';' -f 1`Sort the lines based on process names using `sort` to have a standard order before comparisonUse `tee` to print the resultant strings to the terminal and simultaneously save to a file
Now, let’s apply these transformations to the fresh_run.log as well as resumed_run.log entries.
- →`fresh_run.log`
- →`resumed_run.log`
Inference from process top-level hashes
Computing a hash is a multi-step process and various factors contribute to it such as the inputs of the process, platform, time-stamps of the input files and more ( as explained in Demystifying Nextflow resume blog post) . The change we made in the task level CPUs directive and script section of the FASTQC process triggered a re-computation of hashes:
Even though we only introduced changes in FASTQC, the MULTIQC process was re-run since it relies upon the output of the FASTQC process. Any task that has its cache hash invalidated triggers a rerun of all downstream steps:
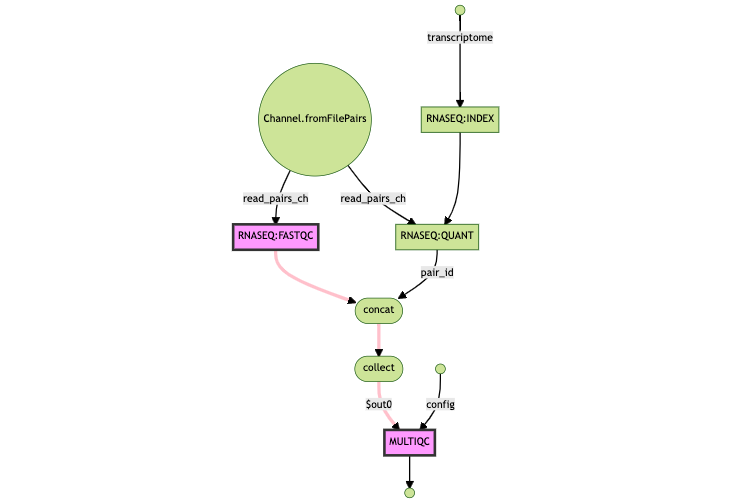
Understanding why `FASTQC` was re-run
We can see the full list of FASTQC process hashes within the fresh_run.log file
When we isolate and compare the log entries for FASTQC between fresh_run.log and resumed_run.log, we see the following diff:
Observations from the diff:
- →We can see that the content of the script has changed, highlighting the new `$task.cpus` part of the command.There is a new entry in the `resumed_run.log` showing that the content of the process level directive `cpus` has been added.
In other words, the diff from log files is confirming our edits.
Understanding why `MULTIQC` was re-run
Now, we apply the same analysis technique for the MULTIQC process in both log files:
Here, the highlighted diffs show the directory of the input files, changing as a result of FASTQC being re-run; as a result MULTIQC has a new hash and has to be re-run as well.
Conclusion
Debugging the caching behavior of a pipeline can be tricky, however a systematic analysis can help to uncover what is causing a particular process to be re-run.
When analyzing large datasets, it may be worth using the -dump-hashes option by default for all pipeline runs, avoiding needing to run the pipeline again to obtain the hashes in the log file in case of problems.
While this process works, it is not trivial. We would love to see some community-driven tooling for a better cache-debugging experience for Nextflow, perhaps an nf-cache plugin? Stay tuned for an upcoming blog post describing how to extend and add new functionality to Nextflow using plugins.